| dc.contributor.author | Tauber, Maïthé | - |
| dc.contributor.author | Thuilleaux, Denise | - |
| dc.contributor.author | Bieth, Éric | - |
| dc.date.accessioned | 2018-05-30T11:38:15Z | |
| dc.date.available | 2018-05-30T11:38:15Z | |
| dc.date.issued | 2015 | |
| dc.identifier.citation | Tauber, Maïthé ; Thuilleaux, Denise ; Bieth, Éric ; Le syndrome de Prader-Willi en 2015, Med Sci (Paris), 2015, Vol. 31, N° 10 ; p. 853-860 ; DOI : 10.1051/medsci/20153110011 | |
| dc.identifier.issn | 1958-5381 | |
| dc.identifier.uri | http://hdl.handle.net/10608/8696 | |
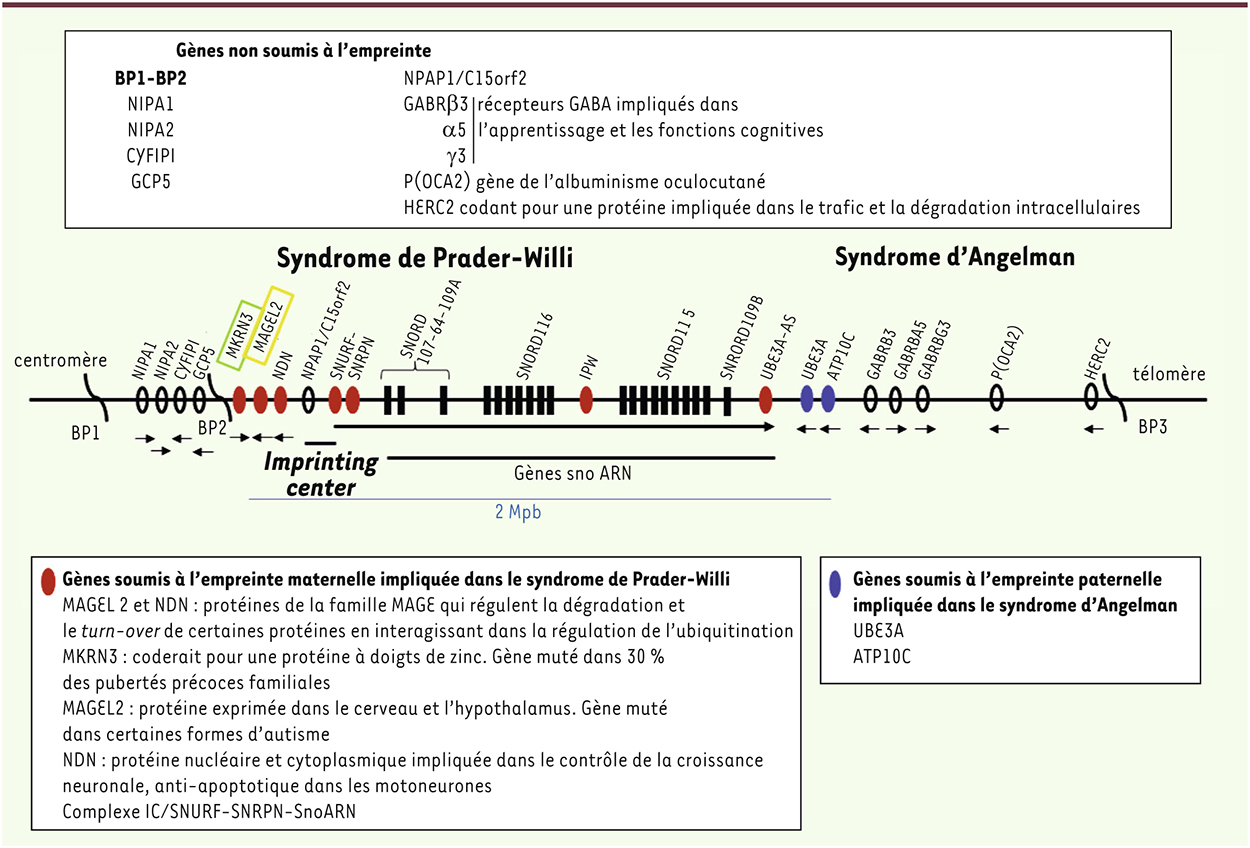
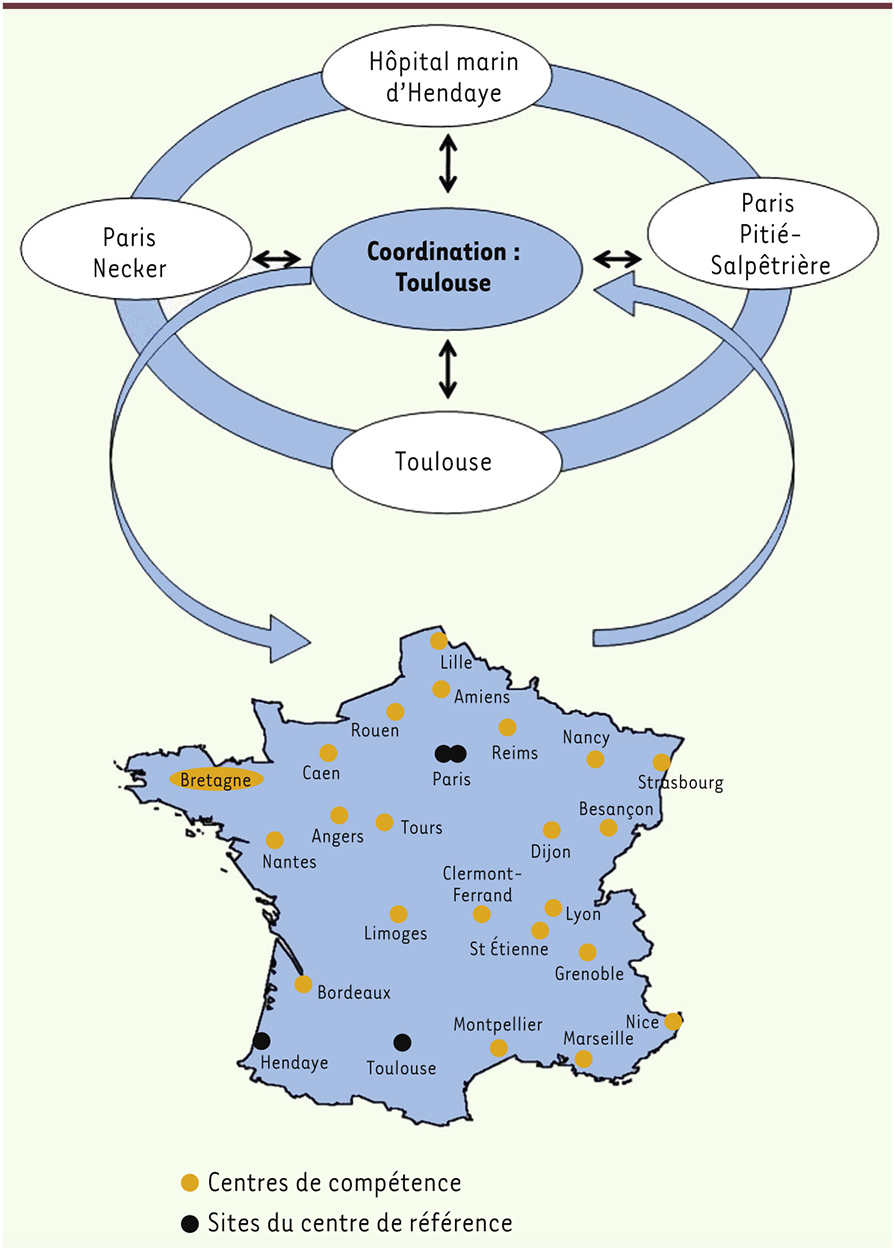
| dc.description.abstract | Le syndrome de Prader-Willi est un trouble du développement lié à un défaut d’expression de gènes de la région 15q11-q12 soumise à une empreinte parentale maternelle. Le diagnostic peut aujourd’hui être porté très tôt, ce qui permet de décrire l’histoire de la maladie et, en particulier, les phases nutritionnelles allant de difficultés alimentaires sévères en période néonatale jusqu’à l’apparition d’une obésité morbide avec hyperphagie et déficit de satiété. S’associent des dysfonctionnements endocriniens, un déficit cognitif modéré, des troubles des apprentissages, des troubles du comportement et des habilités sociales, et des troubles psychiatriques. La prise en charge multidisciplinaire a été fortement développée grâce à la mise en place du plan Maladies rares en 2004 et du plan Obésité en 2011, en lien avec l’association des familles (Prader-Willi France). Des perspectives thérapeutiques physiopathologiques intéressantes existent aujourd’hui. | fr |
| dc.description.abstract | Prader-Willi syndrome is a neurodevelopmental disorder caused by the lack of expression of imprinted genes of the chromosomal region 15q11-q12. Diagnosis can now be made in the first months of life, allowing a precise description of the natural history of the disease. Of interest, nutritional phases appear to be more complex than those initially reported, starting with a severe hypotonia with deficit of suckling and failure to thrive in neonates, and subsequently switching to excessive weight gain with morbid obesity due to hyperphagia and deficit of satiety. The phenotype also includes endocrine dysfunction, intellectual disability, learning deficits, behavioural troubles with impaired social skills and psychiatric features. Multidisciplinary care has been strongly improved by the rare disease programme launched in France in 2004 and the Obesity programme in 2011 in link with the patient association. New therapeutic perspectives have arisen from knowledge of the pathophysiology of PWS. | en |
| dc.language.iso | fr | |
| dc.publisher | Éditions EDK, Groupe EDP Sciences | |
| dc.relation.ispartof | M/S Revues | |
| dc.rights | Article en libre accès | fr |
| dc.rights | Médecine/Sciences - Inserm - SRMS | fr |
| dc.source | M/S. Médecine sciences [ISSN papier : 0767-0974 ; ISSN numérique : 1958-5381], 2015, Vol. 31, N° 10; p. 853-860 | |
| dc.subject.mesh | Diagnostic différentiel | fr |
| dc.subject.mesh | Évolution de la maladie | fr |
| dc.subject.mesh | France | fr |
| dc.subject.mesh | Dépistage génétique | fr |
| dc.subject.mesh | Humains | fr |
| dc.subject.mesh | Hypogonadisme | fr |
| dc.subject.mesh | Troubles mentaux | fr |
| dc.subject.mesh | Phénotype | fr |
| dc.subject.mesh | Syndrome de Prader-Willi | fr |
| dc.subject.mesh | méthodes | fr |
| dc.subject.mesh | diagnostic | fr |
| dc.subject.mesh | génétique | fr |
| dc.subject.mesh | psychologie | fr |
| dc.subject.mesh | complications | fr |
| dc.subject.mesh | thérapie | fr |
| dc.title | Le syndrome de Prader-Willi en 2015 | fr |
| dc.title.alternative | Prader-Willi syndrome in 2015 | en |
| dc.type | Article | |
| dc.contributor.affiliation | Centre de référence du SPW, unité d’endocrinologie, obésité, maladies osseuses, génétique et gynécologie médicale, hôpital des enfants, CHU de Toulouse, 330 avenue de Grande-Bretagne, TSA 70034, 31059 Toulouse Cedex 9, France | |
| dc.contributor.affiliation | Unité Prader-Willi, hôpital marin de Hendaye, Hendaye, France | |
| dc.contributor.affiliation | Génétique médicale, hôpital Purpan, CHU de Toulouse, France | |
| dc.identifier.doi | 10.1051/medsci/20153110011 | |
| dc.identifier.pmid | 26481024 | |