| |
| Med Sci (Paris). 34: 39–41. doi: 10.1051/medsci/201834s211.15es JSFM : Prix communication affichée 2017 Relations génotype-phénotype des mutations du gène de la filamine C (FLNC) Flavie Ader,1* Eric Villard,2 Céline Ledeuil,3 Philippe Charron,4 and Pascale Richard1 1Unité Fonctionnelle de Cardiogénétique et Myogénétique, Centre de Génétique, Hôpitaux Universitaires Pitié Salpêtrière - Charles Foix, Paris, France 2Sorbonne Université, UPMC Univ. Paris 06, Inserm, UMR_S 1166 and ICAN Institute for Cardiometabolism and Nutrition, Paris, France 3Unité Fonctionnelle de Cardiogénétique et Myogénétique, Centre de Génétique, Hôpitaux Universitaires Pitié Salpêtrière - Charles Foix, ParisFrance 4APHP, Centre de référence pour les maladies cardiaques héréditaires, Hôpital Pitié-Salpêtrière, Paris, France |
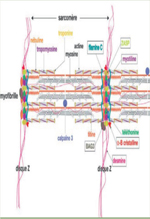
La filamine C est une protéine homodimérique codée par le gène FLNC (7q32, 47 exons ; NM_001458.4), qui joue un rôle structural majeur en interagissant avec les protéines du disque Z, les sarcoglycanes de la membrane cellulaire et l’actine cytosolique. La filamine C est impliquée dans la myogenèse et joue un rôle de senseur au niveau du disque Z participant ainsi à la maintenance et à la réparation des sarcomères [1]. Elle est constituée d’un domaine de liaison à l’actine en N-terminal, de deux domaines ROD1 et ROD2, et d’un domaine de dimérisation en C-terminal. Initialement, des mutations de la filamine C ont été rapportées dans les myopathies autosomiques dominantes myofibrillaires (MMF) et distales (MD). Plus récemment, ce gène a été impliqué dans des cardiomyopathies isolées (cardiomyopathie hypertrophique -CMH-, dilatée -CMD- et restrictive ‑CMR‑). Concernant les myopathies, des mécanismes pathogéniques ont été en revanche décrits [2] dans les cardiomyopathies isolées, la pathophysiologie reste non élucidée. Afin d’explorer l’existence d’une possible relation entre les variants identifiés et le sous-type morphologique de cardiomyopathie, le phénotype de 30 patients atteints de cardiomyopathies et mutés dans le gène FLNC a été étudié et confronté au génotype. |
L’analyse d’un panel de 46 gènes connus de cardiomyopathie a permis d’identifier 30 patients (neuf CMD, 17 CMH et quatre CMR) porteurs d’un variant pathogène dans le gène FLNC. La pathogénicité est définie en fonction des critères de Richards et al. [3] adaptés au regard de la prévalence et des caractéristiques génétiques de la maladie ainsi que de l’expérience du laboratoire. Seuls les variants certainement pathogènes (variants connus, publiés avec des données fonctionnelles ou des variants avec une ségrégation familiale contributive) et probablement pathogènes (nouveaux variants avec une fréquence allélique < 0,01% dans GnomAD et des sites de prédictions de pathogénicité - SIFT, Polyphen, etc. - en faveur d’un effet pathogène) ont été retenus pour l’analyse. |
Résultats cliniques Les 17 patients avec CMH comprenaient 10 cas familiaux et sept cas sporadiques. L’hypertrophie apparaît modérée (15,8 ± 3,0 mm) et des troubles de conduction sont à noter chez 6/17 patients (35 %). Dans le groupe des patients CMD (n = 9), nous avons rapporté quatre cas familiaux et cinq cas sporadiques. Trois patients sont décédés de mort subite (33 %). Enfin dans le groupe des CMR, trois cas sur quatre étaient des formes familiales, avec une sévérité objectivée par deux décès et une transplantation cardiaque chez des adultes jeunes. Pour aucun d’entre eux, il n’est fait mention d’atteinte myopathique, ni d’histoire familiale de myopathie (Tableau I).
Tableau I.
|
CMH (n = 17) |
CMD (n = 9) |
CMR (n = 4) |
| Âge moyen au diagnostic (ans) |
33 [9-66] |
32 [0-51] |
36 [23-46] |
|
| FEVG moyen (%) |
Normal |
37 ± 6 % |
Normal + Profil restrictif |
|
| Épaisseur du septum interventriculaire (mm) |
15,8 ± 3,0 |
NA |
NA |
|
| Épaisseur de la paroi postérieure (mm) |
18,5 ± 4,4 |
NA |
NA |
|
| Appareillage (DAI/PM) |
3/17 |
2/9 |
1/4 |
|
| Histoire de mort subite personnelle ou familiale |
2/17 |
3/9 |
¾ patients décédés ou en attente de greffe |
|
| Anomalies ECG
*FA
*Troubles conduction |
4/17
6/17 |
1/9
2/9 |
1/4 |
|
| CK |
normales |
normales |
normales |
Résumé des données cliniques. |
Relation phénotype-génotype Trente variants dont 19 faux-sens, neuf variants nuls, et deux insertions et délétions en phase ont été retenus. Dix-sept des variants faux-sens sont associés à une CMH et deux à une CMR. Tous les variants nuls ont été identifiés chez des patients atteints de CMD. Les deux insertions et délétions en phase sont retrouvées chez des patients atteints de CMR. Les variants faux-sens sont répartis dans les domaines ROD2 (n = 10) et ROD1 (n = 7) de la protéine alors que les variants nuls sont distribués sur tout le gène. Chez les patients avec CMR, les variants sont retrouvés dans le ROD2 et dans le domaine de dimérisation (Figure 1).
 | Figure 1. Représentation schématique de la filamine C avec les différents domaines (actin binding, ROD1, Hinge1 [H1], ROD2, domaine de dimérisation [D]). Les régions R10 ET R11 sont contiguës. Les variants publiés (HGMD Pro) et ceux de cette cohorte (soulignés) sont positionnés le long de la protéine, en rouge variant identifié chez un patient atteint de CMD, en fuschia variant identifié chez un patient atteint de CMH, en noir variant identifié chez un patient atteint de CMR, en bleu, variant identifié chez un patient atteint de MD et en vert variant identifié chez un patient atteint de MMF. Les variants tronquants sont représentés sur la partie supérieure de la filamine C et les variants non tronquants sur la partie inférieure. |
|
Ce travail a évalué, sur une cohorte de 30 patients présentant des mutations FLNC, la relation génotype-phénotype dans les sous-types de cardiomyopathies. Il apparait que les variants faux-sens sont retrouvés dans les CMH alors que les variants nuls sont toujours retrouvés dans les CMD. Dans la littérature, la prévalence des mutations FLNC dans les CM isolées est de l’ordre de 4 % dans les CMD et de 2 % dans les CMH. Parmi 45 variants publiés dans les cardiomyopathies et répondant à nos critères de validation, 19 sont associés à une CMH, 24 à une CMD, et deux à une CMR. En accord avec notre cohorte, dans le phénotype de CMH, les variants responsables sont majoritairement des faux-sens avec une régionalisation dans le domaine ROD2 alors que dans le phénotype de CMD, ce sont des variants tronquants qui sont identifiés. Si les variants nuls retrouvés dans les CMD agiraient selon un mécanisme d’haplo-insuffisance [4], le mécanisme d’action des variants faux-sens est mal connu (effet dominant négatif). Dans les myopathies, six variants associés à des MD et huit associés à des MMF ont été publiés avec des mutations de tous types. Le nombre de cas rapportés de myopathies liées à des mutations FLNC reste faible et limite les corrélations génotype-phénotype ainsi que l’estimation de la prévalence de ce gène dans le phénotype. Toutefois, les variants du domaine de liaison à l’actine seraient plutôt associés à des MD et ceux du domaine de dimérisation à des MMF [2]. Des atteintes cardiaques ont été rapportées dans 1/3 des MMF associées à une mutation non-sens [5, 6]. |
En conclusion, dans les cardiomyopathies, une corrélation semble émerger entre la nature de la mutation et le sous type de cardiomyopathie. Dans les myopathies, la nature du variant ne semble pas influencer le développement d’un phénotype particulier mais les corrélations génotype-phénotype restent limitées du fait de leur faible prévalence. Dans tous les cas, l’identification d’une mutation du gène FLNC, doit conduire à une prise en charge adaptée sur le plan cardiaque. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
|
1.
Dalkilic I,
Schienda J,
Thompson TG,
Kunkel LM.
Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure . Mol Cell Biol. 2006; ; 26 : :6522.–6534. 2.
Fürst DO,
Goldfarb LG,
Kley RA.
et al. Filamin C-related myopathies: pathology and mechanisms . Acta Neuropathol. 2013; ; 125 : :33.–46. 3.
Richards S,
Aziz N,
Bale S.
et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology . Genet Med. 2015; ; 17 : :405.–424. 4.
Begay RL,
Tharp CA,
Martin A.
et al. FLNC gene splice mutations cause dilated cardiomyopathy . JACC Basic Transl Sci. 2016; ; 1 : :344.–359. 5.
Kley RA,
Hellenbroich Y,
van der Ven PF.
et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients . Brain. 2007; ; 130 : :3250.–3264. 6.
Avila-Smirnow D,
Gueneau L,
Batonnet-Pichon S.
et al. Cardiac arrhythmia and late-onset muscle weakness caused by a myofibrillar myopathy with unusual histopathological features due to a novel missense mutation in FLNC . Rev Neurol (Paris). 2016; ; 172 : :594.–606. |


