I. Définitions, repérage et diagnostic de la déficience intellectuelle
2016
3-
Étiologies environnementales et génétiques
L’identification de l’étiologie d’une déficience intellectuelle (DI) est une question primordiale car elle seule permet d’apporter des éléments de réponse au « pourquoi » des parents, d’optimiser la prise en charge des patients, de ne pas passer à côté d’une cause curable et enfin, d’évaluer le risque de transmission dans la fratrie et la famille. Mais l’extrême hétérogénéité clinique et génétique des déficiences intellectuelles rend ce travail difficile et souvent infructueux. Si une DI est toujours la conséquence d’un événement qui perturbe le développement cérébral soit avant la naissance (prénatal), soit en période périnatale soit durant l’enfance (postnatal), les causes possibles sont multiples et peuvent être d’origines environnementales ou génétiques acquises ou héréditaires.
Causes environnementales
Les causes environnementales représentent environ 15 à 20 % des étiologies des DI. Elles sont regroupées ici en fonction de la période de survenue.
Causes prénatales
Il s’agit de causes intervenant durant la période comprise entre le premier jour des dernières règles et le début du travail d’accouchement.
Embryo-fœtopathies : l’exemple de l’infection à CMV en cours de grossesse
Les infections fœtales dues au cytomégalovirus (CMV), au toxoplasme, aux virus de l’herpès et de la rubéole, etc., augmentent considérablement le risque de déficit neurologique postnatal (OR=2,97 ; IC 95 % [1,52-5,80]) (Badawi et coll., 1998

; Benoist et coll., 2008
; Mwaniki et coll., 2012
). Des mécanismes multiples interviennent dans la cytopathogénicité : arrêt de synthèse cellulaire, accumulation de matériel viral dans la cellule, formation de syncytia par fusion membranaire, induction des processus apoptotiques, etc.
Le CMV est la cause la plus fréquente d’infection congénitale et touche 0,3 à 0,5 % des naissances en Europe de l’Ouest. En France, l’incidence des primo-infections chez les femmes enceintes est de 0,3 à 1,4 %. Le risque de transmission au fœtus dépend essentiellement du statut maternel vis-à-vis du CMV : en cas de primo-infection maternelle, le risque de contamination fœtale est d’environ 30 à 50 %. Ce taux chute à 0,2-2 % lors d’une réinfection. L’âge gestationnel, au moment de la virémie fœtale, n’influence pas le taux de transmission, mais est corrélé à la sévérité de l’infection, les atteintes au cours de la première moitié de la grossesse ayant le plus mauvais pronostic (Stagno et coll., 1986
; Hassan et Connell, 2007
; Lombardi et coll., 2010
).
Environ 5 à 10 % des enfants infectés in utero naissent avec des anomalies cliniques : le nouveau-né est hypotrophe, présente un tableau ictéro-hémorragique (hépatosplénomégalie, pétéchies, purpura), il peut être microcéphale et présenter des calcifications intracrâniennes ou des dilatations ventriculaires. Environ 10 à 20 % des nouveau-nés présentant ce tableau décèdent. Les survivants présentent des séquelles neurosensorielles (retard psychomoteur, surdité uni- ou bilatérale, cécité, retard de langage). Cependant, la grande majorité des enfants infectés ne présente aucune symptomatologie à la naissance, mais 5 à 15 % d’entre eux développeront une surdité neurosensorielle, une choriorétinite et une DI. Ces séquelles sont plus fréquentes, plus graves et d’apparition plus précoce chez les enfants infectés lors d’une primo-infection maternelle que lors d’une seconde infection.
Alors que les conséquences des infections par le virus de la rubéole et par le toxoplasme sont prévenues par le dépistage systématique des séroconversions, inscrit dans la loi, la question du dépistage systématique des séroconversions maternelles à CMV n’est pas encore recommandée. L’infection à CMV peut être détectée dans le sang maternel par une mesure du taux d’anticorps anti-CMV. Cependant, ce test ne permet pas de préciser la date de l’infection, ce qui, en cas de résultat positif, ne permet pas de différencier une infection ancienne (sans risque particulier) d’une primo-infection, dangereuse pour le fœtus. De plus, aucun traitement permettant de prévenir les effets délétères du virus sur le fœtus n’a aujourd’hui fait ses preuves.
Intoxications durant la grossesse : l’exemple du syndrome d’alcoolisation fœtale
L’exposition
in utero à certaines substances tels que des médicaments (certains anti-épileptiques), l’alcool, le tabac, certaines drogues (cocaïne), ou à d’autres produits neurotoxiques (arsenic, mercure, plomb…) peut altérer le développement du système nerveux central (Liu et coll., 2010
; Clifford et coll., 2012
). L’ensemble de ces intoxications serait responsable d’environ 2 % des cas de DI.
L’exemple emblématique est celui de l’alcool. L’alcool traverse aisément la barrière placentaire et les concentrations d’éthanol dans le liquide amniotique et chez le fœtus atteignent des valeurs comparables à celles mesurées dans le sang maternel. Par ailleurs, le taux en éthanol dans le lait maternel est de 10 % supérieur à celui dans le sang (Inserm, 2001
). Une exposition prénatale à l’alcool peut altérer le développement de tous les organes mais l’éthanol exerce ses principaux effets sur le système nerveux central avec parfois de lourdes conséquences sur le développement psychomoteur de l’enfant. Les manifestations les plus graves de l’exposition prénatale à l’alcool portent le nom de syndrome d’alcoolisation fœtale. Son incidence est estimée entre 0,5 et 3 pour 1 000 naissances. Les effets tératogènes de l’alcool se traduisent par des malformations craniofaciales (de petites fentes palpébrales, un étage moyen de la face plat, un petit nez retroussé, un philtrum convexe lisse et long et une lèvre supérieure fine), un retard de croissance et des troubles cognitifs et du comportement. Le risque d’accoucher d’un enfant de faible poids ou d’un enfant présentant des troubles cognitifs apparaît généralement pour des consommations supérieures ou égales à 20 g par jour. Néanmoins, d’après les études expérimentales, il n’a jamais été mis en évidence de dose seuil en deçà de laquelle la consommation d’alcool serait sans danger pour le fœtus.
Les nombreuses observations chez l’Homme et les études réalisées dans des modèles expérimentaux ont permis de mieux comprendre les mécanismes de la toxicité de l’alcool sur le cerveau en développement (Miranda, 2012
). Au cours du développement cérébral, l’éthanol modifie la prolifération neuronale, il induit également une mort neuronale excessive. De plus, l’éthanol interfère avec diverses étapes de la gliogénèse cérébrale, en particulier, il induit une transformation précoce de la glie radiaire, qui joue un rôle clé dans le développement neuronal, constituant un support dans la migration neuronale au niveau du néocortex ; un tel effet de l’éthanol pourrait être la base des ectopies neuronales et d’autres anomalies corticales souvent décrites chez les enfants de mères consommant de l’alcool. Au niveau moléculaire, l’éthanol interagit avec les récepteurs au glutamate de type N-méthyl-D-aspartate et avec les récepteurs au GABA, deux types de récepteurs clefs dans le développement et le fonctionnement cérébral. L’éthanol induit également un stress oxydatif et interfère avec divers facteurs trophiques. Enfin, des données récentes suggèrent que l’éthanol pourrait altérer la fonction de protéines induites dans la réponse cellulaire au stress (cf. communication « Exposition prénatale à l’alcool et troubles causés par l’alcoolisation fœtale » de S. Toutain dans la partie I de cet ouvrage).
Encore insuffisamment évalué, le tabac serait responsable de déficits intellectuels et de troubles du comportement. Il est cependant parfois difficile de dissocier le rôle propre de ces expositions
in utero de celui des facteurs psychosociaux maternels qui sont souvent associés (Braun et coll., 2009
).
Plus récemment, de nombreuses études ont révélé l’effet délétère des polybromodiphényléthers ou PBDE (molécules synthétiques, retardateurs de flamme, ajoutées comme ignifugeants dans les plastiques, les textiles et les équipements électriques et électroniques) sur le neurodéveloppement de l’enfant. Ces composés bromés, suspectés d’être des perturbateurs endocriniens, seraient également responsables de troubles de développement du système nerveux (autisme, hyperactivité, déficit d’attention, trouble de comportement…). Plusieurs études ont montré l’action de ces retardateurs de flamme bromés sur les hormones thyroïdiennes (Ibhazehiebo et coll., 2011a
et b
) et ont établi une corrélation entre l’exposition prénatale et un retard de langage et un trouble de l’attention associés à une réduction de 5,5 à 8 points du QI verbal pour ceux qui avaient eu la plus forte exposition prénatale (Herbstman et coll., 2010
).
Prématurité
La grande prématurité augmente de façon importante le risque de troubles cognitifs. Ce point a été abordé en détails dans un précédent rapport d’expertise collective de l’Inserm (Inserm, 2004
) et ne sera donc pas rediscuté. Les résultats de la cohorte française Epipage sont également présentés dans le chapitre « Repérage et dépistage ».
Pathologies maternelles
Une plus forte prévalence des malformations congénitales et de déficience intellectuelle (Behrooz et coll., 2011
; Bath et coll., 2013
) est observée en présence de certaines carences nutritionnelles durant la période périconceptionnelle. Deux exemples de telles carences sont décrits ci-dessous.
• Hypothyroïdisme, carence en iode
La grossesse entraîne d’importantes modifications de la fonction thyroïdienne maternelle, susceptibles de favoriser l’apparition d’une hypothyroïdie ou son aggravation, particulièrement en cas de carence iodée ou d’auto-immunité. Au cours de la grossesse, les besoins en iode augmentent d’environ 50 μg/j, en raison d’une augmentation de la clairance rénale de l’iode, du transfert fœtoplacentaire de l’iode et d’une stimulation de la thyroïde maternelle (Zimmermann, 2012
).
De par leur rôle dans la neurogenèse, la migration neuronale, la myélinisation et la régulation de la neurotransmission, les hormones thyroïdiennes sont essentielles au développement cérébral du fœtus et du nouveau-né.
La synthèse des hormones thyroïdiennes par la thyroïde fœtale débute vers la 20
e semaine de gestation, aussi les besoins du fœtus au cours des deux premiers trimestres de grossesse sont assurés par la thyroïde maternelle grâce au passage transplacentaire. De ce fait, une hypothyroïdie maternelle affecte le développement neurologique du nouveau-né et de l’enfant (Henrichs et coll., 2013
). De nombreuses études ont montré que l’hypothyroïdie au cours du premier trimestre de grossesse était associée à un faible score développemental des enfants durant la première année de vie, à un QI global inférieur de 7 points chez les enfants nés de mères hypothyroïdiennes non diagnostiquées pendant la grossesse en comparaison aux enfants témoins. Le risque de difficultés d’apprentissage scolaire est multiplié par quatre chez les enfants de mères ayant une hypothyroïdie gestationnelle mal contrôlée (Haddow et coll., 1999
; Smit et coll., 2000
; Grosse et coll., 2011
).
• Hyperphénylalaninémie maternelle
L’hyperphénylalaninémie maternelle est une forme rare de phénylalaninémie qui résulte d’une anomalie du métabolisme d’un acide aminé (la phénylalanine) (voir également paragraphe sur les erreurs innées du métabolisme). Faute d’enzyme adaptée, la phénylalanine ne peut être détruite et son taux augmente anormalement dans le sang et les urines. La phénylcétonurie résultante peut être compensée par un régime alimentaire adapté.
En cas de grossesse, l’excès de phénylalanine dans le sang maternel est un puissant agent tératogène pour l’embryon et le fœtus. C’est la raison pour laquelle les enfants nés de mères phénylcétonuriques, dont le régime a été élargi à l’adolescence, présentent une fœtopathie caractérisée par une DI associée à une microcéphalie, un retard de croissance intra-utérin et une dysmorphie faciale rappelant le syndrome d’alcoolisation fœtale (Prick et coll., 2012
). Le retard de développement psychomoteur est d’autant plus sévère que la phénylalaninémie maternelle est élevée. La phénylalanine a un effet toxique tout au long de la grossesse, avec en début de gestation, des perturbations de l’organogenèse responsables des diverses malformations, puis des troubles de la croissance fœtale avec microcéphalie et retard de croissance intra-utérin. Les atteintes cérébrales retrouvées chez ces enfants montrent des anomalies de la myélinisation identiques à celles retrouvées chez les patients phénylcétonuriques. En l’absence de régime correct, les risques sont maximaux ; en revanche, avec un régime préconceptionnel et une phénylalaninémie normalisée, la croissance fœtale et le développement psychomoteur ultérieur sont normaux, sans malformations.
Devant tout enfant présentant un retard mental associé à une microcéphalie, il est absolument indispensable de rechercher une phenylcétonurie maternelle pour, le cas échéant, mettre en place le traitement adéquat et prévenir les risques de récurrence. Même si en France, le dépistage couvre tous les nouveau-nés depuis 1968, il faut être vigilant pour les mères nées avant cette date et pour celles originaires de pays où ce dépistage n’est pas effectué ou a été mis en place plus tardivement.
Causes péri- et postnatales
Le terme périnatal est généralement réservé à la période se situant entre le début du travail et le 7e jour de vie, le terme postnatal couvrant la période ultérieure au 7e jour de vie.
Au cours de ces périodes, des accidents périnatals (anoxie cérébrale, hémorragie cérébrale, ictère nucléaire…) ou des maladies infectieuses de la petite enfance (méningite, encéphalite, encéphalopathies post-vaccinales) peuvent survenir et être cause de DI.
Ces deux points ont été abordés en détails dans un précédent rapport d’expertise collective de l’Inserm (Inserm, 2004
) ainsi que dans le chapitre « Repérage et dépistage ».
Intoxication : l’exemple des intoxications au plomb
De nombreuses observations chez l’Homme et des études menées sur des modèles animaux ont démontré l’effet neurotoxique d’une exposition à des métaux lourds comme le plomb ou le mercure (Mendola et coll., 2002
; McDermott et coll., 2011
). L’exemple choisi est celui de l’exposition au plomb.
La toxicité du plomb est connue depuis l’antiquité et le saturnisme aigu touchait autrefois principalement les mineurs et les ouvriers de la métallurgie du plomb, ceux qui utilisaient de la vaisselle de plomb et les ouvriers sertissant au plomb, les vitraux. Avec l’avènement de la peinture au plomb et de l’essence plombée, le saturnisme, devenu très courant aux xixe et xxe siècles, fut l’une des six premières maladies à avoir été déclarée maladie professionnelle en octobre 1919.
Si le taux de saturnisme a considérablement décru parmi les professionnels, cette maladie affecte d’autres populations, en particulier la population infantile des habitats anciens par l’ingestion de plomb sous forme de particule fines ou d’écailles de peinture au plomb. Le système nerveux est particulièrement vulnérable aux effets toxiques du plomb, notamment chez l’enfant où une exposition, même à de faibles doses, peut entraîner des anomalies du développement psychomoteur. En témoignent, chez les enfants intoxiqués, la présence de troubles de l’attention et la diminution des performances intellectuelles. Ainsi, une étude menée sur 278 enfants afro-américains vivant en milieu urbain a démontré la toxicité du plomb y compris à faible dose (5 μg/dL) avec une diminution de 5 à 25 points du QI selon le niveau d’exposition (Min et coll., 2009
).
En France, une expertise collective de l’Inserm a estimé en 1999, que 85 000 enfants de 1 à 6 ans étaient encore victimes de ce type de saturnisme (Inserm, 1999
). En 2006, ce sont encore 437 nouveaux cas de saturnisme infantile qui ont été repérés en métropole. Cependant, une étude de prévalence menée par l’Institut de veille sanitaire (InVS) en 2008 et 2009 montre que le nombre d’enfants de 1 à 6 ans atteints par le saturnisme a été divisé par 20 entre 1995 et 2008.
Outre ses effets sur la production de l’acide delta aminolévulinique déshydratase (ALAD), une protéine essentielle à la biosynthèse de l’hème, élément constitutif de l’hémoglobine, le plomb interfère également avec certains systèmes de neurotransmission. Plusieurs actions pharmacologiques ont été décrites : liaison avec les protéines, action sur la libération de certains neurotransmetteurs (glutamate, dopamine), action sur les canaux calciques voltage-dépendants et ceux liés au récepteur glutamatergique de type N-méthyl-D-aspartate (NMDA), action sur la protéine C et sur le métabolisme énergétique de la mitochondrie. Enfin, des effets délétères du plomb sur la différenciation astrocytaire ont également été observés. Le plomb altère donc la majorité des mécanismes neurobiologiques essentiels du développement cérébral.
Traumatisme crânien
Le traumatisme crânien est une atteinte cérébrale ou bulbaire provoquée par le contact brusque (accélération, décélération ou rotation) entre le tissu cérébral et la boîte crânienne.
Le traumatisme crânien de l’enfant est fréquent, le plus souvent causé par des accidents de la voie publique (60 % des cas), des chutes (25 %) ou des accidents de sport pour les 15 % restants. Il est la première cause de mortalité chez les moins de 15 ans et l’une des principales causes de handicap avec de multiples séquelles cognitives. Après un traumatisme crânien sévère, une perte d’une quinzaine de points de QI, en moyenne, est observée chez l’enfant. Des résultats récents montrent un QI inférieur de 18 à 26 points 10 ans après un traumatisme sévère survenu chez des enfants de 2 à 7 ans par rapport au groupe contrôle (Anderson et coll., 2012
). Parallèlement, d’autres fonctions cognitives sont également altérées : la mémoire à court et à long terme, de travail et la mémoire prospective. Enfin, les troubles de l’attention sont mal évalués et souvent sous-estimés alors qu’ils sont très gênants.
Dans tous les cas, la gravité des séquelles cognitives est corrélée à l’ampleur du traumatisme crânien.
Facteurs psychosociaux et économiques (malnutrition, abus, carences émotionnelles)
Plusieurs études convergent pour suggérer un impact des facteurs psychosociaux et économiques (stress maternel, statut socio-économique de la famille, manque de cohésion sociale, maltraitance) sur la survenue d’un déficit intellectuel. Pour se développer au mieux, les enfants ont besoin de trouver dans leur entourage, parental ou autres, un soutien émotionnel et une stimulation cognitive. Contrairement aux facteurs organiques, les facteurs psychosociaux apparaissent d’autant plus importants qu’on se situe dans le cadre de la déficience légère (cf. également chapitre « Prévalences des déficiences intellectuelles »).
Par exemple, une étude finlandaise a établi une corrélation positive entre le niveau d’instruction des parents, leur statut socio-économique et la prévalence de DI (Heikura et coll., 2008
). De même, une étude américaine a montré que des enfants nés de mères avec moins de 12 années de scolarisation ont un risque 7 fois plus élevé de présenter un retard cognitif comparé à ceux nés de mères ayant suivi des études supérieures (Chapman et coll., 2002
). Une autre étude américaine a montré l’effet protecteur significatif d’un niveau élevé d’éducation maternelle (de plus de 13 années ou niveau bac) (Bilder et coll., 2013
).
Enfin, il est important de noter que, même pour les DI de cause organique déterminée, les conditions environnementales, psychologiques et sociales jouent un rôle dans l’aggravation ou l’atténuation des troubles comme l’atteste, par exemple, la différence du devenir des prématurés, sans lésion neurologique, en fonction des caractéristiques sociales et psychologiques du milieu familial (Ment et coll, 1996
; Thompson et coll., 1997
; Vohr et coll., 2003
).
Causes génétiques
On estime qu’environ 1/3 des 25 000 gènes humains sont exprimés au niveau du cerveau, participant à son développement et fonctionnement. C’est sans doute l’une des explications de l’extrême hétérogénéité génétique des déficiences intellectuelles, l’altération de l’un ou l’autre de ces gènes pouvant affecter le développement cognitif.
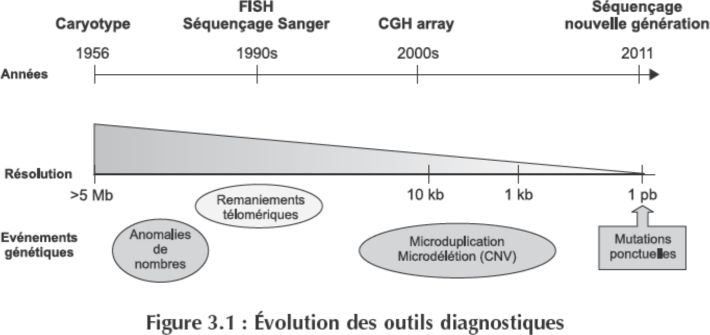
Tous les modes de transmission mendéliens (autosomique ou lié au chromosome X, dominant ou récessif) et non mendéliens (c’est-à-dire sans altération de la séquence du génome nucléaire) sont décrits. Comme le montre la figure 3.1
, le développement d’outils de diagnostic de plus en plus performants a permis au cours des 60 dernières années, un gain en sensibilité d’un facteur 1 000 000.
Anomalies chromosomiques
Les anomalies chromosomiques sont l’étiologie la plus fréquente de la DI. Elles sont observées dans 0,7 % des naissances vivantes, et concernent environ 16 % des DI sévères et 5 % des DI légères (Raymond et Tarpey, 2006
; Ellison et coll, 2013
). Il s’agit de modifications du nombre de chromosomes par perte ou gain d’un chromosome complet (aneuploïdie), ou d’anomalies de structures telles que des délétions, duplications, dérivés de translocations ou d’inversions. L’aneuploïdie est un phénomène fréquent lors de la conception et on estime à 25 % le taux de blastocystes présentant une formule chromosomique anormale (Regan et Willatt, 2010
). Les DI causées par les anomalies de structures sont la plupart du temps syndromiques et caractérisées par des atteintes multisystémiques. C’est la raison pour laquelle Schmickel introduit dès 1986 la notion de « syndrome de gènes contigus » pour expliquer l’ensemble des signes cliniques caractéristiques d’une microdélétion hémizygote
1
Un organisme est dit hémizygote pour un gène ou groupe de gènes particuliers si ces derniers ne sont présents qu’à une seule copie (c’est-à-dire présents sur un seul allèle) à un locus donné dans un organisme diploïde.
. Il propose que le phénotype observé chez ces patients résulte de l’haploinsuffisance de l’ensemble des gènes contenus dans le segment délété. Si cette théorie a été contredite pour certaines pathologies où l’anomalie d’un seul gène suffit à entraîner le phénotype (syndrome d’Angelman ou de Smith-Magenis par exemple), elle reste vraie dans la très grande majorité des cas.
Anomalies de nombre
Par définition, les anomalies de nombre affectent le nombre des chromosomes et non leur structure. Suivant le stade du développement embryonnaire où elles se produisent, ces anomalies peuvent être retrouvées de façon homogène, dans toutes les cellules de l’organisme, ou en mosaïque, c’est-à-dire que dans certaines cellules de l’individu. Lorsqu’elles sont homogènes, elles résultent le plus souvent d’une non-disjonction méiotique lors de la gamétogenèse parentale et peuvent se traduire par une trisomie (présence d’un chromosome normal surnuméraire) ou par une monosomie (perte d’un chromosome). Lorsqu’elles sont mosaïques, elles résultent d’une non-disjonction mitotique, survenue dans les premiers stades du développement de l’embryon.
Le syndrome de Down (SD ou trisomie 21) est la forme génétique la plus fréquente de DI. Le QI de ces enfants est extrêmement variable, compris entre 20 et 80 avec une valeur moyenne à 50. Outre le retard intellectuel, ce syndrome se caractérise par des malformations (surtout cardiaques, plus rarement digestives ou rénales) et une dysmorphie faciale. Diverses études ont montré que 90 % des trisomies 21 résultent d’une erreur au cours de la méiose maternelle et 8 % d’une erreur paternelle. Dans 2 % des cas, il existe une non-disjonction mitotique postzygotique (Visootsak et Sherman, 2008
; Turleau et Vekemans, 2010
). La probabilité d’avoir un enfant trisomique augmente avec l’âge de la mère.
L’étude des trisomies 21 partielles (seule une partie du chromosome 21 est en excédent) et de modèles murins, combinée aux connaissances récentes sur la carte physique et le transcriptome, a permis d’identifier plusieurs gènes candidats du chromosome 21 directement ou indirectement impliqués dans la pathogénicité du syndrome de Down. De nombreuses études visent à décrire les voies métaboliques contrôlées par ces gènes en vue de nouvelles stratégies thérapeutiques. Ainsi, le gène
DYRK1A code une sérine/thréonine kinase dont l’activité contrôle de nombreuses voies de signalisation intracellulaires (NOTCH, NFAT, P53) (Sitz et coll., 2008
; Lu et coll., 2011
; Mazur-Kolecka et coll., 2012
). Il existe un inhibiteur naturel de cette enzyme, un puissant antioxydant extrait du thé vert, le gallate d’épigallocatéchine ou EGCG. Un premier essai clinique a débuté en 2010 (De la Torre et coll., 2014
).
Outre la trisomie 21, les anomalies autosomiques observables à la naissance et associées à une DI sont les trisomies 13 et 18. Décrites en 1960, ces anomalies sont extrêmement sévères et rapidement létales. Elles sont en général diagnostiquées in utero en raison du tableau malformatif dépistable à l’échographie. Lorsque la trisomie 13 est présente en mosaïque (car survenue dans les premiers stades du développement et donc ne concernant qu’une partie des cellules), elle peut entraîner des tableaux cliniques variables sur le plan malformatif et neurodéveloppemental. Il n’existe pas de corrélation entre le taux de la mosaïque et le pronostic vital ou mental, ce qui rend difficile le conseil génétique en cas de diagnostic prénatal de mosaïque.
Anomalies de structures
Les anomalies de structure résultent de cassures chromosomiques suivies par un ou plusieurs recollements anormaux. Les anomalies de structure peuvent affecter un chromosome ou plusieurs chromosomes, homologues ou non. Elles peuvent être équilibrées ou non équilibrées. Les anomalies équilibrées n’entraînent généralement pas de déséquilibre du matériel chromosomique et n’ont donc pas d’effet phénotypique sauf si la cassure interrompt un gène ou altère son expression. Cependant, les anomalies équilibrées peuvent entraîner, lors de la méiose, la formation de gamètes déséquilibrés donnant des zygotes anormaux, ce qui se traduira par la survenue d’avortements ou par la naissance d’enfants porteurs d’anomalies congénitales. Les anomalies non équilibrées peuvent survenir de novo ou être la conséquence d’un remaniement parental équilibré.
• Remaniements télomériques
Les régions télomériques sont particulièrement riches en gènes, mais aussi en séquences minisatellites hypervariables (HVP) qui facilitent les cassures. Les séquences répétées en tandem de petites tailles (< 60 pb) composant ces structures favorisent l’apparition d’erreurs d’alignement (mésappariement), conduisant à des crossing-over inégaux. Ce mécanisme aboutit à l’élimination d’une région sur l’un des chromosomes et à la duplication sur l’autre chromosome. Ces événements submicroscopiques échappent à une étude cytogénétique de routine.
En 1995, en étudiant la transmission de régions polymorphiques présentes au niveau des télomères, Flint et coll. ont mis en évidence 5 % d’anomalies chromosomiques cryptiques chez des patients présentant une DI syndromique (Flint et coll., 1995
). Deux ans plus tard, un premier panel de sondes explorant les régions subtélomériques était développé permettant de détecter 7,5 % d’anomalies dans la même population de patients (Knight et coll., 1997
). Au cours des années suivantes, la description phénotypique détaillée de patients porteurs de ces remaniements a permis l’individualisation de syndromes cliniquement reconnaissables comme les délétions 1p36 ; 2q37 (ostéodystrophie héréditaire d’Albright type 3) ; 3pter ; 9q34 (syndrome Kleefstra) ; 18qter et 22q13 (syndrome Phelan McDermid). À la fin des années 1990, les remaniements télomériques représentaient la cause la plus fréquente de DI après la trisomie 21.
• Microdélétions, duplications
Dans les années 2000, l’avènement de la technique de l’analyse comparative sur puce à ADN (
CGH array pour
Comparative Genomic Hybridization array) a permis de déceler deux fois plus d’anomalies chromosomiques chez les patients avec DI, mettant en évidence des remaniements submicroscopiques (appelés CNV pour
Copy Number Variant), non visibles sur caryotype, car de taille inférieure à 5 Mb
2
Mb : mega base pairs (106 paires de base).
. Les progrès considérables de cette technique (jusqu’à 2 millions de sondes sur les dernières puces CGH) permettent de visualiser des remaniements aussi petits que 10 kb
3
kb : kilo base pairs (103 paires de base).
de taille.
Plusieurs anomalies chromosomiques de petite taille sont récurrentes. Les données issues du séquençage du génome permettent d’expliquer cette récurrence qui est liée à l’architecture du génome. En effet, ces anomalies sont médiées par des structures génomiques appelées duplicons ou LCR (Low Copy Repeats). Ces répétitions segmentaires ayant une grande homologie de séquence (95-99 %), de 10 kb à 400 kb de longueur, sont présentes en plusieurs copies dans le génome, espacées de 500 kb à 4 Mb. Elles sont particulièrement présentes dans les régions centromériques et télomériques. Lors de la méiose, la grande homologie de séquence de ces blocs d’ADN favorise les recombinaisons homologues non-alléliques illégitimes (NAHR), conduisant à une délétion ou duplication de la région comprise entre deux duplicons de même orientation. Lorsque ce mécanisme est à l’origine d’un déséquilibre chromosomique, la taille du segment remanié est toujours identique entre les patients parce que les points de cassure se situent dans les duplicons. Le phénotype clinique de ces désordres génomiques résulte vraisemblablement d’un dosage anormal des gènes localisés dans le fragment génomique remanié. En fonction de la taille du fragment génomique impliqué, ces désordres génomiques peuvent se manifester sous la forme de syndrome de gènes contigus ou de maladie mendélienne. Enfin, les déséquilibres aboutissant à des excès de dosages géniques (duplications, trisomies) sont généralement moins délétères que ceux aboutissant à des déficits (délétions, monosomies).
Le tableau 3.I
résume les principaux syndromes de DI causés par des CNV récurrents et cliniquement reconnaissables (Stankiewicz et Lupski, 2010
).
Classiquement, les syndromes ont été initialement décrits cliniquement (comme la trisomie 21) et les bases génétiques élucidées secondairement. La technique de CGH array a conduit à la démarche inverse avec l’identification de l’anomalie chromosomique précédant la description phénotypique. En effet, la recherche systématique d’anomalies chromosomiques chez les patients présentant une DI idiopathique a permis, par l’analyse clinique rétrospective des patients, de décrire de nouveaux syndromes associés à des microdélétions ou microduplications. La reconnaissance clinique de ces syndromes favorise ensuite une recherche ciblée du remaniement chez d’autres patients par des techniques de FISH (Fluorescence In Situ Hybridization) ou de biologie moléculaire (PCR quantitative). Plusieurs dizaines de syndromes ont été décrits à ce jour, la majorité de ces syndromes ayant une prévalence faible, de l’ordre de 1/10 000.
En pratique, la technique de CGH array de résolution moyenne (400 kb) est aujourd’hui l’un des piliers de l’évaluation diagnostique de l’enfant atteint d’une DI syndromique. Les nombreuses études publiées à ce jour montrent que cet examen révèle une anomalie chromosomique chez environ 7 à 20 % des patients présentant une DI syndromique.
Tableau 3.I Principaux syndromes neurogénétiques et CNV (Copy Number Variants) associés
|
Syndrome
|
OMIM*
|
Locus
|
Réarrangement
|
Gène(s)
|
|
Williams Beuren Syndrome del(7)q11.23
|
194050
|
7q11.23
|
Délétion
|
Syndrome de gènes contigus incluant ELN
|
|
dup(7)q11.23
|
609757
|
7q11.23
|
Duplication
|
Syndrome de gènes contigus incluant ELN
|
|
Angelman Syndrome
|
105830
|
15q11–q12
|
Délétion maternelle, disomie uniparentale paternelle 15
|
UBE3A
|
|
Prader-Willy Syndrome
|
76270
|
15q11–q12
|
Délétion paternelle, disomie uniparentale maternelle 15
|
Syndrome de gènes contigus
|
|
dup(15)
|
608636
|
15q11–q13
|
Duplication
|
Syndrome de gènes contigus
|
|
Miller-Dieker Syndrome
|
247200
|
17p13.3
|
Délétion
|
Syndrome de gènes contigus incluant LIS1
|
|
Smith-Magenis Syndrome
|
182290
|
17p11.2
|
Délétion
|
Syndrome de gènes contigus incluant RAI1
|
|
del(17)q21.31
|
610443
|
17q21.31
|
Délétion
|
Syndrome de gènes contigus incluant KANSL1
|
|
DiGeorge/Velo-Cranio-Facial Syndrome
|
188400
|
22q11.2
|
Délétion
|
Syndrome de gènes contigus incluant TBX1, COMT
|
|
del(22)q13
|
606232
|
22q13.3
|
Délétion
|
SHANK3/PROSAP2
|
|
Pelizaeus-Merzbacher Syndrome
|
312080
|
Xq22.2
|
Duplication, délétion
|
PLP1
|
* OMIM : Online Mendelian Inheritance in Man, base de données « on line » qui répertorie toutes les maladies connues à composantes génétiques
Abréviations : ELN : Elastin gene ; UBE3A : Ubiquitin protein ligase E3A ; LIS1 : Isolated Lissencephaly Sequence ; RAI1 : Retinoic Acid Induced 1 ; KANSL1 : KAT8 regulatory NSL complex subunit 1 ; TBX1 : T-box 1 ; COMT : Catechol-O-methyltransferase ; SHANK3 : SH3 and multiple ankyrin repeat domains 3 ; PROSAP2 : Proline-rich synapse-associated protein 2
Déficiences intellectuelles à mode de transmission mendélien
DI liées au chromosome X (DILX)
L’histoire des DILX commence plus d’un siècle avant l’avènement de la cytogénétique et l’ère moléculaire : durant un siècle (1868-1968), un certain nombre d’entités cliniques (maladies de Pelizaeus-Merzbacher, la dystrophie musculaire Duchenne, l’incontinentia pigmenti ou syndrome de Bloch-Sulzberger, des erreurs innées de métabolisme ou syndrome de Hunter) et de grands pedigrees dans lesquels la DI ségrégeait selon un mode lié au chromosome X, ont été englobés. Pendant la même période, plusieurs études rapportaient l’excès de garçons parmi des personnes avec DI.
Ces deux observations – l’excès de garçons parmi des personnes avec DI et des syndromes cliniques ségrégant selon un mode lié à l’X – suggéraient une contribution importante des anomalies géniques du chromosome X à l’étiologie de ces affections. En 1943, J. Purdon Martin et Julia Bell décrivirent un arbre généalogique de retard mental lié à l’X (Martin et Bell, 1943
). En 1969, Herbert Lubs décrivit pour la première fois un chromosome X inhabituel associé au retard mental (Lubs, 1969
). En 1970, Frédérick Hecht introduit le terme de site fragile. Mais il a fallu attendre 1991-1992 pour l’identification du gène responsable de la fragilité (
FMR1), de sa protéine FMRP (
Fragile Mental Retardation Protein), et du type de mutation (dite mutation dynamique ou instable) à l’origine de ce syndrome par des équipes françaises et internationales.
Plus récemment, la recherche sur l’étiologie des DILX a bénéficié d’une part de l’optimisation des technologies de séquençage (plusieurs dizaines de gènes étudiées chez un grand nombre de malades) et d’autre part, de stratégies collaboratives s’appuyant sur des réseaux de cliniciens et de chercheurs. Ainsi, le réseau européen EuroMRX, créé en 1996, a apporté une contribution majeure à ces travaux. Grâce aux études de liaison génétique qui ont pu être conduites sur de grandes cohortes de familles de patients, à la constitution d’une base de données cliniques détaillées et de nombreuses études moléculaires, près d’une vingtaine de gènes de DILX ont été identifiés par ce consortium (de Brouwer et coll., 2007
). De même, de nouveaux gènes de DILX (Raymond et coll., 2009
; Tarpey et coll., 2009
) ont été identifiés grâce à l’étude IGOLD (
International Genetics of Learning Disability) de reséquençage des exons codants de 718 gènes du chromosome X, sur une cohorte de 208 familles de DILX.
À ce jour, les DILX regroupent plus de 150 syndromes dans lesquels la DI apparaît comme la caractéristique première (Lubs et coll., 2012
) avec 102 gènes responsables décrits. La très grande majorité des cas de DILX est causée par des mutations ponctuelles inactivant ces gènes, et environ 10 % des cas de DILX seraient liés à de petites anomalies (délétion ou duplication) touchant ces gènes.
Parmi ces DILX, le syndrome de l’X fragile (FXS) est la deuxième cause la plus fréquente des DI (1 %) et représente la première cause monogénique. Cette maladie génétique rare est associée à un déficit intellectuel léger à sévère pouvant être associé à des troubles du comportement et à des signes physiques caractéristiques. Sa prévalence estimée est d’environ 1/2 500 naissances (prévalence de la mutation complète) à 1/4 000 (cas symptomatiques) dans les deux sexes.
Le FXS est dû à l’inactivation transcriptionnelle du gène
FMR1 (
Fragile X Mental Retardation 1), localisé en Xq27.3 : l’expansion d’une répétition de triplets d’acides nucléiques (CGG)n, dans la région 5’ non traduite de ce gène aboutit à un nombre de répétitions supérieur à 200, et favorise des méthylations qui répriment l’expression du gène (Verkerk et coll., 1991
).
Ces mutations dites complètes proviennent d’allèles instables, prémutés (55
à 200 répétitions de CGG). De rares cas de mutations ponctuelles intragéniques ont été décrits dans
FMR1 sans expansion de répétitions CGG (Penagarikano et coll., 2007
). Le gène
FMR1 code pour FMRP, une protéine de liaison aux ARNs, impliquée dans diverses étapes du métabolisme des ARN messagers (Ashley et coll., 1993
). En absence de FMRP, on observe la surexpression de protéines importantes pour la structure et la fonction synaptique provoquant l’altération de la dépression à long terme (LTD) et de la morphologie des épines dendritiques, et perturbant la plasticité synaptique (Bassell et Warren, 2008
). L’absence de FMRP cause également une up-régulation du récepteur à glutamate métabotropique sub-type 5 (mGlu5). Cette observation est à l’origine de plusieurs essais cliniques utilisant des antagonistes de ces récepteurs (Bear et coll., 2004
).
Pour les autres gènes de DILX, l’ensemble des travaux démontre que chacun des gènes ne rend compte que d’un très petit nombre de cas, ce qui pose le problème de la mise en place de leur diagnostic moléculaire. De plus, au-delà de la démonstration de l’extrême hétérogénéité génétique des DILX, ces travaux révèlent également l’extrême variabilité phénotypique intra- et interfamiliale des mutations de ces gènes. Un même phénotype peut résulter d’altération de gènes différents du chromosome X et, inversement, un même gène peut occasionner différents phénotypes. Des gènes connus comme responsables de formes syndromiques de DI peuvent ainsi être également impliqués dans des déficiences mentales non ou peu spécifiques.
Formes autosomiques dominantes (DIAD)
La déficience intellectuelle est fréquemment retrouvée dans les syndromes polymalformatifs. Dans l’immense majorité des cas, ces syndromes surviennent de façon sporadique. Cette prépondérance de cas sporadiques suggère la survenue d’une mutation hétérozygote
de novo. Cependant, devant l’extrême hétérogénéité clinique et probablement génétique de ces syndromes, peu d’études ont tenté d’établir leurs bases génétiques. C’est grâce à la cytogénétique que les premiers gènes de DIAD ont été identifiés. Il s’agissait d’analyser les points de cassure de translocations apparemment équilibrées à phénotype anormal, ou de séquencer les gènes contenus dans des micro-délétions chez un grand nombre de patients ayant une DI (
SHANK2 ou
ARID1B par exemple) (Berkel et coll., 2010
; Hoyer et coll., 2012
). Toutefois, seuls quelques gènes ont pu être identifiés par ces approches.
Les développements technologiques en matière de séquençage de l’ADN ont permis de nouvelles avancées sur les connaissances étiologiques génétiques des DIAD. Le projet canadien «
Synapse to Disease » (S2D), initié en 2006, a été le premier à utiliser une stratégie de séquençage systématique de gènes candidats. Il portait sur l’analyse de 500 gènes codant des protéines synaptiques dans une cohorte comprenant 95 sujets avec DI non-syndromiques, 142 autistes non-syndromiques, 134 schizophrènes et 190 individus contrôles. Ceci a permis l’identification, chez des patients ayant une DI non syndromique, de mutations délétères survenues
de novo dans des gènes codant des protéines du système glutamatergique :
SYNGAP1, STXBP1, SHANK3, KIF1A, GRIN1, CACNG2, EPB41L1 (Hamdan et coll., 2009
et 2011
).
Depuis 5 ans, la révolution génomique a conduit au développement de nouveaux outils de séquençage à très haut-débit. Si la dernière génération des séquenceurs à capillaires, utilisant la technique Sanger, permettait de lire jusqu’à 2 millions de bases en une demi-journée, de nouvelles machines dotées de débits de 50 à 1 000 fois supérieurs sont apparues sur le marché en 2007. Ces séquenceurs de nouvelle génération ont permis de s’affranchir d’un certain nombre de biais de la méthode Sanger comme la nécessité de cloner l’ADN à séquencer. C’est grâce notamment à la lecture de plusieurs millions de séquences en parallèle que ces nouveaux séquenceurs à « haut débit » ont pu révolutionner les analyses en génomique en permettant, entre autre, le re-séquençage massif de tout ou partie d’un génome pour en identifier les variations.
Même si les régions régulatrices de l’ADN, les microARN et autres éléments modulateurs suscitent aujourd’hui beaucoup d’intérêt, il n’en reste pas moins que la majorité des mutations impliquées dans des maladies génétiques mendéliennes a été trouvée dans les régions codantes. L’exome, qui correspond à l’ensemble des exons codant du génome et représente 1 % ou 2 % de notre ADN, semble donc particulièrement intéressant à analyser pour identifier de nouveaux gènes responsables de maladies génétiques. L’intérêt d’une approche combinée d’enrichissement par hybridation et de séquençage d’exome pour identifier des mutations rares dans des pathologies humaines est aujourd’hui largement démontré (Ng et coll., 2010
; Ku et coll., 2011
). Ces dernières années ont vu le développement de plusieurs projets très ambitieux visant à séquencer l’exome de dizaines de patients présentant une déficience intellectuelle et de leurs parents sains afin d’identifier l’ensemble des mutations survenues
de novo chez ces patients (Vissers et coll., 2010
; de Ligt et coll., 2012
; Rauch et coll., 2012
). Plusieurs informations essentielles résultent de ces travaux. Tout d’abord, ils indiquent qu’environ 20 % des DI ont une origine génétique autosomique dominante. Comme observé dans les formes liées au chromosome X, les résultats attestent de l’extrême hétérogénéité génétique de ces anomalies et du très petit nombre de patients souffrant de mutations d’un même gène. Enfin, ils apportent une nouvelle démonstration de la variabilité de l’expression clinique des mutations de gènes de DI.
Formes autosomiques récessives (DIAR)
Bien qu’un effort considérable ait été consenti pour l’étude des gènes du chromosome X, il n’en reste pas moins que les formes autosomiques récessives sont considérées, comme de beaucoup, les plus fréquentes. Elles représenteraient environ un quart des cas de DI.
• Erreurs innées du métabolisme
Parmi les formes de DI d’hérédité récessive autosomique, le groupe des erreurs innées du métabolisme (EIM) occupe une place prépondérante. Les EIM sont des maladies génétiques caractérisées par la dysfonction d’une enzyme ou d’une protéine impliquée dans le métabolisme cellulaire. Elles constituent environ un tiers des maladies génétiques d’étiologie connue et peuvent toucher tous les organes. Plus de 400 maladies différentes sont connues qui ont, chez l’enfant, une incidence globale entre 1/2 000 et 1/4 000. La majorité de ces déficits entraîne l’absence d’un composé situé en aval de la voie biochimique ainsi bloquée et/ou l’accumulation d’un composé situé en amont du déficit enzymatique. Un désordre cellulaire en découle soit par carence (anomalie de biosynthèse), soit par intoxication par accumulation d’un des composés. Entités nombreuses, mais chacune étant rare, leur présentation clinique est très polymorphe. Elles sont responsables d’une DI rarement isolée et/ou fixée. Atteinte auditive, visuelle, viscérale, squelettique et des signes neurologiques comme une régression des acquisitions, ataxie, convulsions, mouvements anormaux et troubles du comportement sont souvent présents. Toutefois, l’anomalie de certaines voies métaboliques peut être responsable d’une DI fixée, non syndromique ou associée à des signes cliniques peu spécifiques, comme la phénylcétonurie, le déficit en créatine, le défaut de synthèse des purines, l’acidurie 4-hydroxybutyrique (Garcia-Cazorla et coll., 2009
).
Le diagnostic des maladies métaboliques est crucial car, en dehors du conseil génétique, il autorise un traitement dans un grand nombre de cas. Il peut s’agir soit d’un traitement diététique (suppression d’un composé toxique pour le patient, apport calorique, évitement du jeûne selon les cas), soit de l’apport d’un cofacteur qui ne peut être synthétisé ou qui peut pallier le déficit enzymatique, soit enfin d’une enzymothérapie apportant l’enzyme manquante.
• Phénylcétonurie
L’exemple classique de maladie d’intoxication est celui de la phénylcétonurie (PCU) car la plus commune des EIM. Elle est caractérisée par un déficit mental léger à sévère chez les patients non traités. Elle fut décrite pour la première fois par Asbjorn Folling en 1934 à l’école de médecine de l’Université d’Oslo chez 2 enfants présentant une DI et une présence d’acide phénylpyruvique dans les urines. Sa prévalence a une variabilité géographique considérable. Elle est d’environ 1/10 000 naissances en Europe (1/17 000 en France ; 1/4 000 en Turquie) (Blau et coll., 2010
).
Cette affection est due au déficit d’une enzyme hépatique, la phénylalanine-4-hydroxylase (PAH), qui catalyse la transformation de la phénylalanine contenue dans l’alimentation en tyrosine. Les dysfonctionnements de la PAH conduisent à une accumulation de phénylalanine dans l’organisme, atteignant des niveaux toxiques en particulier dans le cerveau.
La relation entre l’apport de phénylalanine et la PCU fut soulignée pour la première fois, en 1954 par le professeur allemand Horst Bickel. Il établit qu’un régime pauvre en phénylalanine chez une fillette de 3 ans chez qui le diagnostic de phénylcétonurie avait été tardif, avait permis de normaliser le taux sanguin de phénylalanine et d’améliorer la capacité mentale et l’état général de cette patiente (Bickel et coll., 1953
). Depuis le début des années 1960, un dépistage systématique de la PCU est réalisé chez les nouveau-nés grâce au test de Guthrie, afin d’introduire un régime alimentaire dès les premiers jours de vie et d’éviter une détérioration mentale irréversible. Les enfants chez qui le taux de phénylalanine est maintenu tout au long de leur vie, dans les normes conseillées, auront un développement neurologique normal.
• Anomalies de la biosynthèse de la créatine
Les anomalies de la biosynthèse de la créatine regroupent 3 maladies qui provoquent une carence en créatine (Stockler et coll., 2007
; Braissant et coll., 2011
).
La créatine, synthétisée dans le foie et le pancréas à partir de l’arginine, passe dans le sang pour être véhiculée vers les muscles, le cœur, le cerveau et d’autres tissus où elle est phosphorylée par la créatine kinase pour être stockée sous forme de phosphocréatine. Cette forme phosphorylée constitue une source de phosphates pour la synthèse de l’ATP dans les cellules cérébrales et musculaires. Les déficits de deux enzymes intervenant dans la synthèse de la créatine, la guanidinoacétate méthyltransférase (GAMT) et l’arginine glycine amidinotransférase (AGAT) ainsi que celui du récepteur de la créatine (Xq28) sont à la source des trois maladies responsables de handicap intellectuel. Cette DI peut être isolée ou associée à un autisme, à un syndrome extra-pyramidal ou à une épilepsie. La créatinine plasmatique dosée par le ionogramme sanguin peut être diminuée ou normale selon la méthode biochimique utilisée. Les dosages de guanidinoacétate et de créatine dans les urines et le plasma orientent le diagnostic de façon précise. Les dosages enzymatiques se font à partir de lymphocytes, tissu hépatique ou fibroblastes. L’imagerie anatomique cérébrale est normale, le plus souvent, ou peut parfois montrer une anomalie de signal des globi pallidi. La spectroscopie IRM, quant à elle, montre l’absence de pic de créatine dans tous les cas, même en cas de défaut du récepteur. Ainsi la spectroscopie IRM, qui devrait idéalement être réalisée avec toute IRM cérébrale dans l’investigation étiologique d’un retard psychomoteur, permet le diagnostic des déficits en créatine.
Les déficits en GAMT et AGAT sont traités de façon efficace par la créatine orale (350 mg/kg/jour-2 g/kg/jour) alors que le déficit en transporteur ne semble pas répondre aux traitements par créatine, arginine et lysine (précurseurs de la créatine). Un régime pauvre en arginine et riche en ornithine est également prescrit dans le déficit en GAMT du fait d’une possible toxicité cérébrale du guanidinoacétate (Valayannopoulos et coll., 2012
). Cependant, l’administration de créatine peut parfois suffire à diminuer le taux de guanidinoacétate car la créatine exerce un contrôle négatif sur l’enzyme AGAT.
• Autres formes récessives
Des avancées majeures ont été réalisées dans le domaine des DIAR, notamment non syndromiques, grâce au développement d’outils génétiques performants comme la méthode de cartographie par autozygotie (ou homozygotie par filiation) et l’analyse de familles consanguines. La consanguinité fait
émerger de nombreuses pathologies récessives et augmente de manière significative le risque d’avoir un enfant avec une DI de 0,012 pour un couple aléatoire à 0,062 pour un couple de cousins germains (Bashi, 1977
; Morton, 1978
). La méthode de cartographie par autozygotie, décrite en 1987 par Lander et Bostein
, consiste à utiliser des familles consanguines « multiplex » (dans de telles familles, tous les sujets atteints sont homozygotes par descendance non seulement pour la mutation morbide mais aussi pour les marqueurs avoisinants) pour localiser des gènes de maladies autosomiques récessives rares. Les sujets atteints homozygotes par descendance pour l’allèle porteur de la maladie, ont hérité de deux copies identiques de l’allèle muté présent en simple exemplaire chez l’ancêtre commun. Les malades sont également homozygotes par descendance pour le fragment de chromosome bordant le locus morbide. D’autres régions sont également homozygotes par descendance, mais sont différentes d’un individu atteint à l’autre. Cette stratégie a donc pour but d’identifier des régions homozygotes communes aux enfants atteints et génodifférentes des enfants sains (homozygotes sauvages ou hétérozygotes). L’efficacité de cette méthode dépend de deux paramètres importants : le nombre total d’enfants dans la famille et le degré de consanguinité ; plus celui-ci est éloigné, plus le nombre de recombinaisons augmente et plus la région suspecte est réduite. L’étude se poursuit alors par l’analyse de gènes candidats qui consiste à étudier les gènes présents dans l’intervalle critique et dont le dysfonctionnement pourrait rendre compte du phénotype observé. C’est grâce à cette stratégie que le premier gène impliqué dans une DIAR non syndromique a été identifié en 2002 (Molinari et coll., 2002
). Une dizaine de gènes a ensuite été identifiée par la même stratégie (Raymond et Tarpey, 2006
; Basel-Vanagaite, 2007
; Ropers, 2010
; Ellison et coll., 2013
).
Plus récemment, la possibilité de combiner l’approche de cartographie par autozygotie à la puissance des outils de séquençage à très haut débit a très nettement favorisé l’identification des gènes responsables de DIAR. L’étude irano-allemande de Ropers et coll. (Najmabadi et coll., 2011
) illustre bien les forces mais aussi les faiblesses de cette nouvelle approche d’analyse dans le domaine des DI. Une étude de liaison utilisant la cartographie par autozygotie a été d’abord menée chez 138 familles consanguines atteintes de DIAR, afin d’identifier les régions homozygotes. Dans un second temps, une puce de capture couvrant l’ensemble des régions candidates a été développée pour le séquençage de la totalité de ces régions chez le probant (personne atteinte de la maladie génétique dans la famille) de chaque famille. Chez 78 familles, l’analyse a identifié une variation prédite pathogène pouvant expliquer la pathologie. Chez 26 familles parmi les 78, des mutations ont été retrouvées dans des gènes déjà connus pour être responsables de DIAR principalement syndromiques (par exemple :
WDR62, SRD5A3, AHI). Les 50 autres variants sont autant de nouveaux gènes candidats de DIAR. Cependant, en l’absence d’autres patients et de toute analyse fonctionnelle, l’imputabilité de ces variants au DIAR reste non démontrée. Enfin, 37 familles présentent plusieurs mutations prédites pathogènes, suggérant soit que l’association de ces variations est à l’origine de la DI (polygénisme) soit qu’une seule d’entre elles est le variant causal.
Déficiences intellectuelles à mode de transmission non mendélien
Au début des années 1980, des expériences de transfert nucléaire réalisées chez la souris révélèrent l’existence d’un phénomène de marquage des génomes parentaux, appelé empreinte génomique parentale. Par la suite, l’identification de gènes spécifiques soumis à empreinte parentale a permis de montrer que cette empreinte conduit à une expression monoallélique, c’est-à-dire un seul des deux allèles (maternel ou paternel) est actif alors que les deux allèles de la plupart des gènes sont actifs ou inactifs de la même façon. Un gène peut être soumis à empreinte seulement dans un tissu particulier (par exemple uniquement dans le placenta) ou à un moment particulier (par exemple au cours du développement embryonnaire). À ce jour, plus de 100 gènes humains soumis à empreinte ont été caractérisés, mais on estime à environ 1 % la fraction de ces gènes présentant une expression différentielle des 2 allèles. Ces gènes sont le plus souvent regroupés dans des domaines chromatiniens placés sous le contrôle d’un centre d’empreinte appelés DMR (
Differentially Methylated Regions) (Franklin et Mansuy, 2011
).
Les caractéristiques moléculaires de ce phénomène de marquage épigénétique ont maintenant été décrites et permettent d’expliquer certaines maladies humaines liées à des gènes soumis à empreinte (Lim et Maher, 2009
; Chamberlain et Lalande, 2010
). Il s’agit principalement des méthylations de l’ADN, mais aussi des acétylations et méthylations des histones, ces protéines sur lesquelles s’enroule l’ADN pour former la chromatine. Ces maladies sont la conséquence soit de la perte de l’allèle actif (par un mécanisme commun comme une délétion ou un mécanisme particulier comme une unidisomie), soit d’une mutation du centre d’empreinte, soit de l’expression anormale de l’allèle normalement silencieux (« relaxation d’empreinte »). Les syndromes de Prader-Willi et d’Angelman sont les deux exemples emblématiques de DI résultant d’anomalies d’empreinte (Gurrieri et Accadia, 2009
; Buiting, 2010
; Chamberlain et Lalande, 2010
).
Le syndrome de Prader-Willi est caractérisé par une hypotonie sévère pendant la période néonatale, des troubles majeurs du comportement alimentaire (entraînant une obésité sévère et des complications pouvant conduire jusqu’au décès précoce), une petite taille postnatale, un hypogonadisme hypogonadotrophique, des mains et des pieds courts, un visage caractéristique, un retard mental modéré (Cassidy et coll., 2012
). L’incidence du syndrome de Prader-Willi est de l’ordre de 1/15 000 naissances. Cette affection est un syndrome des gènes contigus lié à l’absence ou l’inactivation des allèles paternels actifs d’un ensemble de gènes de la région 15q11-q13 du chromosome 15, non compensés par la présence des allèles maternels inactifs. L’anomalie moléculaire sous-jacente responsable du syndrome peut être soit :
• une délétion de la région 15q11-q13 du chromosome 15 d’origine paternelle pour 70 % des malades ;
• une disomie uniparentale maternelle du chromosome 15 pour 28 % des malades (les deux chromosomes 15 proviennent de la mère) ;
• une mutation du centre d’empreinte pour 2 % des malades. Lorsque le centre d’empreinte est muté, les cellules germinales perdent la possibilité de donner l’épigénotype approprié.
Le syndrome d’Angelman est cliniquement distinct du syndrome de Prader-Willi, bien qu’également lié à des anomalies de la région 15q11-q13 (Van Buggenhout et Fryns, 2009
). Il se caractérise par un retard mental sévère, une absence de langage, des accès de rires inappropriés, une microcéphalie, une ataxie, des convulsions, un EEG caractéristique et parfois une hypopigmentation (certaines délétions). L’incidence est de 1/20 000 naissances. Seule la copie maternelle du gène
UBE3A, codant une ubiquitine ligase, est active dans le système nerveux central, alors que l’expression du gène est biallélique dans tous les autres tissus (Williams et coll., 2010
; Mabb et coll., 2011
). C’est l’absence de contribution maternelle, qui est responsable de cette pathologie.
L’anomalie moléculaire sous-jacente responsable du syndrome peut être soit :
• une délétion de la région 15q11-q13 du chromosome 15 d’origine maternelle pour 70 % des malades ;
• une mutation du gène UBE3A pour 20 % des malades ;
• une disomie uniparentale paternelle est responsable de 5 % des cas (les deux chromosomes 15 proviennent du père) ;
• une mutation du centre d’empreinte dans environ 5 % des cas.
En conclusion, des avancées majeures ont été réalisées au cours des vingt dernières années dans la recherche des étiologies des DI, conduisant à l’identification aujourd’hui des causes dans plus de 50 % des formes sévères et 20 % des formes légères. Cependant, la fréquence élevée de mutations de novo et non récurrentes rend d’autant plus difficile le choix des stratégies diagnostiques.
Ces causes sont très diverses et conduisent à de multiples scénarii physiopathologiques. Dans de rares cas encore, ces avancées ont permis de mettre en place les premiers essais thérapeutiques.
Au-delà de la caractérisation de nouveaux gènes responsables de DI, les défis de demain seront de mieux comprendre l’ensemble des mécanismes physiopathologiques sous-jacents et d’identifier des processus cellulaires communs dans lesquels interviennent les produits de ces gènes, qui pourraient représenter autant de cibles thérapeutiques potentielles.
Bibliographie
[1] anderson v,
godfrey c,
rosenfeld jv,
catroppa c. Predictors of cognitive function and recovery 10 years after traumatic brain injury in young children.
Pediatrics. 2012;
129:e254
-e261

[2] ashley ct jr,
wilkinson kd,
reines d,
warren st. FMR1 protein: conserved RNP family domains and selective RNA binding.
Science. 1993;
262:563
-566
[3] badawi n,
watson l,
petterson b,
blair e,
slee j, et coll. . What constitutes cerebral palsy ?.
Dev Med Child Neurol. 1998;
40:520
-527
[4] basel-vanagaite l. Genetics of autosomal recessive non-syndromic mental retardation: recent advances.
Clin Genet. 2007;
72:167
-174
[5] bashi j. Effects of inbreeding on cognitive performance.
Nature. 1977;
266:440
-442
[6] bassell gj,
warren st. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function.
Neuron. 2008;
60:201
-214
[7] bath sc,
steer cd,
golding j,
emmett p,
rayman mp. Effect of inadequate iodine status in UK pregnant women on cognitive outcomes in their children: results from the Avon Longitudinal Study of Parents and Children (ALSPAC).
Lancet. 2013;
382:331
-337
[8] bear mf,
huber km,
warren st. The mGluR theory of fragile X mental retardation.
Trends Neurosci. 2004;
27:370
-377
[9] behrooz hg,
tohidi m,
mehrabi y,
behrooz eg,
tehranidoost m,
azizi f. Subclinical hypothyroidism in pregnancy: intellectual development of offspring.
Thyroid. 2011;
21:1143
-1147
[10] benoist g,
jacquemard f,
leruez-ville m,
ville y. Cytomegalovirus (CMV) congenital infection.
Gynecol Obstet Fertil. 2008;
36:248
-260
[11] berkel s,
marshall cr,
weiss b,
howe j,
roeth r, et coll. . Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation.
Nat Genet. 2010;
42:489
-491
[12] bickel h,
gerrard j,
hickmans em. Influence of phenylalanine intake on phenylketonuria.
Lancet. 1953;
265:812
-813
[13] bilder da,
pinborough-zimmerman j,
bakian av,
miller js,
dorius jt, et coll. . Prenatal and perinatal factors associated with intellectual disability.
Am J Intellect Dev Disabil. 2013;
118:156
-176
[14] blau n,
van spronsen fj,
levy hl. Phenylketonuria.
Lancet. 2010;
376:1417
-1427
[15] braissant o,
henry h,
beard e,
uldry j. Creatine deficiency syndromes and the importance of creatine synthesis in the brain.
Amino Acids. 2011;
40:1315
-1324
[16] braun jm,
daniels jl,
kalkbrenner a,
zimmerman j,
nicholas js. The effect of maternal smoking during pregnancy on intellectual disabilities among 8-year-old children.
Paediatr Perinat Epidemiol. 2009;
23:482
-491
[17] buiting k. Prader-Willi syndrome and Angelman syndrome.
Am J Med Genet C Semin Med Genet. 2010;
154C:365
-376
[18] cassidy sb,
schwartz s,
miller jl,
driscoll dj. Prader-Willi syndrome.
Genet Med. 2012;
14:10
-26
[19] chamberlain sj,
lalande m. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13.
Neurobiol Dis. 2010;
39:13
-20
[20] chapman da,
scott kg,
mason ca. Early risk factors for mental retardation: Role of maternal age and maternal education.
Am J Ment Retard. 2002;
107:46
-59
[21] clifford a,
lang l,
chen r. Effects of maternal cigarette smoking during pregnancy on cognitive parameters of children and young adults: a literature review.
Neurotoxicol Teratol. 2012;
34:560
-570
[22] de brouwer ap,
yntema hg,
kleefstra t,
lugtenberg d,
oudakker ar, et coll. . Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium.
Hum Mutat. 2007;
28:207
-208
[23] de la torre r,
de sola s,
pons m,
duchon a,
de lagran mm, et coll. . Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans.
Mol Nutr Food Res. 2014;
58:278
-288
[24] de ligt j,
willemsen mh,
van bon bw,
kleefstra t,
yntema hg, et coll. . Diagnostic exome sequencing in persons with severe intellectual disability.
N Engl J Med. 2012;
367:1921
-1929
[25] ellison jw,
rosenfeld ja,
shaffer lg. Genetic basis of intellectual disability.
Annu Rev Med. 2013;
64:441
-450
[26] flint j,
wilkie ao,
buckle vj,
winter rm,
holland aj,
mcdermid he. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation.
Nat Genet. 1995;
9:132
-140
[27] franklin tb,
mansuy im. The involvement of epigenetic defects in mental retardation.
Neurobiol Learn Mem. 2011;
96:61
-67
[28] garcia-cazorla a,
wolf ni,
serrano m,
moog u,
perez-duenas b, et coll. . Mental retardation and inborn errors of metabolism.
J Inherit Metab Dis. 2009;
32:597
-608
[29] grosse sd,
van vliet g. Prevention of intellectual disability through screening for congenital hypothyroidism: how much and at what level ?.
Arch Dis Child. 2011;
96:374
-379
[30] gurrieri f,
accadia m. Genetic imprinting: the paradigm of Prader-Willi and Angelman syndromes.
Endocr Dev. 2009;
14:20
-28
[31] haddow je,
palomaki ge,
allan wc,
williams jr,
knight gj, et coll. . Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child.
N Engl J Med. 1999;
341:549
-555
[32] hamdan ff,
gauthier j,
spiegelman d,
noreau a,
yang y, et coll. . Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation.
N Engl J Med. 2009;
360:599
-605
[33] hamdan ff,
gauthier j,
araki y,
lin dt,
yoshizawa y, et coll. . Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability.
Am J Hum Genet. 2011;
88:306
-316
[34] hassan j,
connell j. Translational mini-review series on infectious disease: congenital cytomegalovirus infection: 50 years on.
Clin Exp Immunol. 2007;
149:205
-210
[35] heikura u,
taanila a,
hartikainen al,
olsen p,
linna sl, et coll. . Variations in prenatal sociodemographic factors associated with intellectual disability: A study of the 20-year interval between two birth cohorts in Northern Finland (English).
Am J Epidemiol. 2008;
167:169
-177
[36] henrichs j,
ghassabian a,
peeters rp,
tiemeier h. Maternal hypothyroxinemia and effects on cognitive functioning in childhood: how and why ?.
Clin Endocrinol (Oxf). 2013;
79:152
-162
[37] herbstman jb,
sjodin a,
kurzon m,
lederman sa,
jones rs, et coll. . Prenatal exposure to PBDEs and neurodevelopment.
Environ Health Perspect. 2010;
118:712
-719
[38] hoyer j,
ekici ab,
endele s,
popp b,
zweier c, et coll. . Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability.
Am J Hum Genet. 2012;
90:565
-572
[39] ibhazehiebo k,
iwasaki t,
kimura-kuroda j,
miyazaki w,
shimokawa n,
koibuchi n. Disruption of thyroid hormone receptor-mediated transcription and thyroid hormone-induced Purkinje cell dendrite arborization by polybrominated diphenyl ethers.
Environ Health Perspect. 2011a;
119:168-
-175
[40] ibhazehiebo k,
iwasaki t,
okano-uchida t,
shimokawa n,
ishizaki y,
koibuchi n. Suppression of thyroid hormone receptor-mediated transcription and disruption of thyroid hormone-induced cerebellar morphogenesis by the polybrominated biphenyl mixture, BP-6.
Neurotoxicology. 2011b;
32:400
-409
[41]inserm. Plomb dans l’environnement. Quels risques pour la santé ?.
Collection Expertise collective. Éditions Inserm;
1999;
[42]inserm. Alcool. Effets sur la santé.
Collection Expertise collective. Éditions Inserm;
2001;
[43]inserm. Déficiences et handicaps d’origine périnatale. Dépistage et prise en charge.
Collection Expertise collective. Éditions Inserm;
2004;
[44] knight sj,
horsley sw,
regan r,
lawrie nm,
maher ej, et coll. . Development and clinical application of an innovative fluorescence in situ hybridization technique which detects submicroscopic rearrangements involving telomeres.
Eur J Hum Genet. 1997;
5:1
-8
[45] ku cs,
naidoo n,
pawitan y. Revisiting Mendelian disorders through exome sequencing.
Hum Genet. 2011;
129:351
-370
[46] lander es,
botstein d. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children.
Science. 1987;
236:1567
-1570
[47] lim dh,
maher er. Human imprinting syndromes.
Epigenomics. 2009;
1:347
-369
[48] liu y,
mcdermott s,
lawson a,
aelion cm. The relationship between mental retardation and developmental delays in children and the levels of arsenic, mercury and lead in soil samples taken near their mother’s residence during pregnancy.
Int J Hyg Environ Health. 2010;
213:116
-123
[49] lombardi g,
garofoli f,
stronati m. Congenital cytomegalovirus infection: treatment, sequelae and follow-up.
J Matern Fetal Neonatal Med. 2010;
233:45
-48
[50] lu m,
zheng l,
han b,
wang l,
wang p, et coll. . REST regulates DYRK1A transcription in a negative feedback loop.
J Biol Chem. 2011;
286:10755
-10763
[51] lubs ha. A marker X chromosome.
Am J Hum Genet. 1969;
21:231
-244
[52] lubs ha,
stevenson re,
schwartz ce. Fragile X and X-linked intellectual disability: four decades of discovery.
Am J Hum Genet. 2012;
90:579
-590
[53] mabb am,
judson mc,
zylka mj,
philpot bd. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes.
Trends Neurosci. 2011;
34:293
-303
[54] martin jp,
bell j. A pedigree of mental defect showing sex-linkage.
J Neurol Psychiatry. 1943;
6:154
-157
[55] mazur-kolecka b,
golabek a,
kida e,
rabe a,
hwang yw, et coll. . Effect of DYRK1A activity inhibition on development of neuronal progenitors isolated from Ts65Dn mice.
J Neurosci Res. 2012;
90:999
-1010
[56] mcdermott s,
wu jl,
cai b,
lawson a,
aelion cm. Probability of intellectual disability is associated with soil concentrations of arsenic and lead.
Chemosphere. 2011;
84:31
-38
[57] mendola p,
selevan sg,
gutter s,
rice d. Environmental factors associated with a spectrum of neurodevelopmental deficits.
Mental Retard Dev Disabil Res Rev. 2002;
8:188
-197
[58] ment lr,
vohr b,
oh w,
scott dt,
allan wc, et coll. . Neurodevelopmental outcome at 36 months’ corrected age of preterm infants in the Multicenter Indomethacin Intraventricular Hemorrhage Prevention Trial.
Pediatrics. 1996;
98:714
-718
[59] min mo,
singer lt,
kirchner hl,
minnes s,
short e, et coll. . Cognitive development and low-level lead exposure in poly-drug exposed children.
Neurotoxicol Teratol. 2009;
31:225
-231
[60] miranda rc. MicroRNAs and fetal brain development: Implications for ethanol teratology during the second trimester period of neurogenesis.
Front Genet. 2012;
3:77
[61] molinari f,
rio m,
meskenaite v,
encha-razavi f,
auge j, et coll. . Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation.
Science. 2002;
298:1779
-1781
[62] morton ne. Effect of inbreeding on IQ and mental retardation.
Proc Natl Acad Sci USA. 1978;
75:3906
-3908
[63] mwaniki mk,
atieno m,
lawn je,
newton cr. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review.
Lancet. 2012;
379:445
-452
[64] najmabadi h,
hu h,
garshasbi m,
zemojtel t,
abedini ss, et coll. . Deep sequencing reveals 50 novel genes for recessive cognitive disorders.
Nature. 2011;
478:57
-63
[65] ng sb,
buckingham kj,
lee c,
bigham aw,
tabor hk, et coll. . Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 2010;
42:30
-35
[66] penagarikano o,
mulle jg,
warren st. The pathophysiology of fragile x syndrome.
Annu Rev Genomics Hum Genet. 2007;
8:109
-129
[67] prick bw,
hop wc,
duvekot jj. Maternal phenylketonuria and hyperphenylalaninemia in pregnancy: pregnancy complications and neonatal sequelae in untreated and treated pregnancies.
Am J Clin Nutr. 2012;
95:374
-382
[68] rauch a,
wieczorek d,
graf e,
wieland t,
endele s, et coll. . Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study.
Lancet. 2012;
380:1674
-1682
[69] raymond fl,
tarpey p. The genetics of mental retardation.
Hum Mol Genet. 2006;
15:R110
-R116
[70] raymond fl,
whibley a,
stratton mr,
gecz j. Lessons learnt from large-scale exon re-sequencing of the X chromosome.
Hum Mol Genet. 2009;
18:R60
-R64
[71] regan r,
willatt l. Mental retardation: definition, classification and etiology.
Genetics of mental retardation. An overview encompassing learning disability and intellectual disability.
In: knightsjl, editors.
Karger;
2010;
[72] ropers hh. Genetics of early onset cognitive impairment.
Annu Rev Genomics Hum Genet. 2010;
11:161
-187
[73] sitz jh,
baumgartel k,
hammerle b,
papadopoulos c,
hekerman p, et coll. . The Down syndrome candidate dual-specificity tyrosine phosphorylation-regulated kinase 1A phosphorylates the neurodegeneration-related septin 4.
Neuroscience. 2008;
157:596
-605
[74] smit bj,
kok jh,
vulsma t,
briet jm,
boer k,
wiersinga wm. Neurologic development of the newborn and young child in relation to maternal thyroid function.
Acta Paediatr. 2000;
89:291
-295
[75] stagno s,
pass rf,
cloud g,
britt wj,
henderson re, et coll. . Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome.
JAMA. 1986;
256:1904
-1908
[76] stankiewicz p,
lupski jr. Structural variation in the human genome and its role in disease.
Annu Rev Med. 2010;
61:437
-455
[77] stockler s,
schutz pw,
salomons gs. Cerebral creatine deficiency syndromes: clinical aspects, treatment and pathophysiology.
Subcell Biochem. 2007;
46:149
-166
[78] tarpey ps,
smith r,
pleasance e,
whibley a,
edkins s, et coll. . A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation.
Nat Genet. 2009;
41:535
-543
[79] thompson rj Jr,
gustafson ke,
oehler jm,
catlett at,
brazy je,
goldstein rf. Developmental outcome of very low birth weight infants at four years of age as a function of biological risk and psychosocial risk.
J Dev Behav Pediatr. 1997;
18:91
-96
[80] turleau c,
vekemans m. Trisomy 21: fifty years between medicine and science.
Med Sci (Paris). 2010;
26:267
-272
[81] valayannopoulos v,
boddaert n,
chabli a,
barbier v,
desguerre i, et coll. . Treatment by oral creatine, L-arginine and L-glycine in six severely affected patients with creatine transporter defect.
J Inherit Metab Dis. 2012;
35:151
-157
[82] vanbuggenhout g,
fryns jp. Angelman syndrome (AS, MIM 105830).
Eur J Hum Genet. 2009;
17:1367
-1373
[83] verkerk aj,
pieretti m,
sutcliffe js,
fu yh,
kuhl dp, et coll. . Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.
Cell. 1991;
65:905
-914
[84] visootsak j,
sherman sl. Trisomy 21: Causes and Consequences.
Int Rev Res Ment Retard. 2008;
36:61
-102
[85] vissers le,
deligt j,
gilissen c,
janssen i,
steehouwer m, et coll. . A de novo paradigm for mental retardation.
Nat Genet. 2010;
42:1109
-1112
[86] vohr br,
o’shea m,
wright ll. Longitudinal multicenter follow-up of high-risk infants: why, who, when, and what to assess.
Semin Perinatol. 2003;
27:333
-342
[87] williams ca,
driscoll dj,
dagli ai. Clinical and genetic aspects of Angelman syndrome.
Genet Med. 2010;
12:385
-395
[88] zimmermann mb. The effects of iodine deficiency in pregnancy and infancy.
Paediatr Perinat Epidemiol. 2012;
261:108
-117
→ Aller vers SYNTHESE