2011
| ANALYSE |
13-
Mécanismes associant stress et pathologies
Ce chapitre traitant des liens entre stress et pathologie n’est pas spécifique aux travailleurs indépendants, les mécanismes physiologiques sous-jacents sont les mêmes que pour les travailleurs salariés, et sont généralisables à toutes les situations de stress chronique. Nous allons ici détailler les mécanismes physiologiques par lesquels un état de stress peut aboutir à des pathologies : comprendre le passage d’un état normal à un état pathologique. En effet, la réponse de stress est une réponse de l’organisme – via la libération de neurotransmetteurs et d’hormones – dont le but est l’adaptation à une situation évaluée comme contraignante. Toutefois, le fonctionnement de ce système de défense peut s’altérer en particulier en cas de sur-sollicitation : on parle alors de stress chronique, état psychophysiologique pouvant aboutir à des pathologies.
L’issue pathologique du processus d’adaptation est le résultat d’une combinatoire complexe de facteurs liés :
• aux caractéristiques de l’environnement ;
• à la réactivité psychobiologique générale de l’individu, à ses possibilités d’action comportementale et à l’efficacité de cette démarche ;
• et enfin aux caractéristiques individuelles du fonctionnement des systèmes biologiques impliqués qui orientent la « sortie » pathologique.
Véritables « maladies de l’adaptation », les pathologies liées au stress chronique ont fait l’objet de nombreuses études épidémiologiques lesquelles ont précisé les relations statistiques entre certaines contraintes de travail et ces altérations de santé. L’importance des pathologies concernées sur la santé publique n’est plus à démontrer. Pour certaines de ces pathologies, les évidences mécanistiques sont solides mais pour la plupart elles restent à établir. Quoiqu’il en soit, les mécanismes mis en jeu par la réponse de stress sont multiples, incluant le système nerveux central, le système nerveux autonome, le système neuroendocrinien et le système immunitaire. Seront évoqués dans ce chapitre les mécanismes pour lesquels les connaissances scientifiques sont suffisamment établies.
Santé mentale
Les données épidémiologiques montrent qu’il existe un lien entre des situations de stress chronique et des altérations de la santé mentale : burnout (Ahola et coll., 2006 ; Twellaar et coll., 2008), troubles de l’humeur (anxiété, dépression) (Godin et coll., 2005 ; Melchior et coll., 2007 ; Netterstrøm et coll., 2008 ; Bonde, 2008 ; Siegrist, 2008), troubles du sommeil (Akerstedt, 2006 ; Armon et coll., 2008), troubles des comportements consommatoires (toxicomanies, alcool) (Head et coll., 2004 ; Siegrist et Rödel, 2006).
; Twellaar et coll., 2008), troubles de l’humeur (anxiété, dépression) (Godin et coll., 2005 ; Melchior et coll., 2007 ; Netterstrøm et coll., 2008 ; Bonde, 2008 ; Siegrist, 2008), troubles du sommeil (Akerstedt, 2006 ; Armon et coll., 2008), troubles des comportements consommatoires (toxicomanies, alcool) (Head et coll., 2004 ; Siegrist et Rödel, 2006).
; Twellaar et coll., 2008), troubles de l’humeur (anxiété, dépression) (Godin et coll., 2005 ; Melchior et coll., 2007 ; Netterstrøm et coll., 2008 ; Bonde, 2008 ; Siegrist, 2008), troubles du sommeil (Akerstedt, 2006 ; Armon et coll., 2008), troubles des comportements consommatoires (toxicomanies, alcool) (Head et coll., 2004 ; Siegrist et Rödel, 2006).Il n’existe pas à ce jour un mécanisme causal unique reliant stress et santé mentale, et la littérature expérimentale à ce sujet est considérablement dense. De nombreuses modalités physiopathologiques sont identifiées grâce à des modèles animaux, quelques uns sont suggérés chez l’homme. Parmi ces modalités, l’axe hypothalamo-hypophyso-surrénalien, ou axe corticotrope, semble largement impliqué. L’hyperactivité de ce système en réponse au stress est très certainement pilotée par une hypersécrétion de Corticotropin-Releasing Hormone (CRH), encore appelée Corticotropin-Releasing Factor (CRF), au niveau hypothalamique. L’activation excessive de l’axe est également responsable de l’augmentation de la sécrétion basale de glucocorticoïdes observée chez certains patients déprimés, probablement due à une déficience du rétrocontrôle de l’axe (Pariante et Lightmann, 2008) (figure 13.1). Des anomalies circadiennes telles des avances de phase du rythme de cortisol et des réductions de l’amplitude des rythmes sont également évoquées comme un des liens possibles reliant stress chronique et dépression (Souêtre et coll., 1989 ; Keller et coll., 2006 ; Wirtz-Justice, 2006). En revanche, la réponse à la question « troubles des rythmes circadiens et dépression : cause ou conséquence ? » n’est pas donnée à ce jour.
) (figure 13.1). Des anomalies circadiennes telles des avances de phase du rythme de cortisol et des réductions de l’amplitude des rythmes sont également évoquées comme un des liens possibles reliant stress chronique et dépression (Souêtre et coll., 1989 ; Keller et coll., 2006 ; Wirtz-Justice, 2006). En revanche, la réponse à la question « troubles des rythmes circadiens et dépression : cause ou conséquence ? » n’est pas donnée à ce jour.Récemment, l’hypothèse d’une altération de la plasticité neuronale (structurelle et fonctionnelle) due au stress chronique et conduisant à la dépression a été affinée ouvrant la voie à de nouvelles thérapeutiques (Fuchs et coll., 2004 ; Pittenger et Duman, 2008). Une exposition prolongée aux glucocorticoïdes semble également impliquée dans les atrophies de l’hippocampe décrites par Sapolsky dès 2000 chez les patients déprimés (Sapolsky, 2000). Dans le même sens, les déficits mnésiques rencontrés chez les dépressifs s’expliqueraient par une atténuation de la potentialisation à long terme1
sous stress chronique (De Kloet, 2004).
; Pittenger et Duman, 2008). Une exposition prolongée aux glucocorticoïdes semble également impliquée dans les atrophies de l’hippocampe décrites par Sapolsky dès 2000 chez les patients déprimés (Sapolsky, 2000). Dans le même sens, les déficits mnésiques rencontrés chez les dépressifs s’expliqueraient par une atténuation de la potentialisation à long terme1
sous stress chronique (De Kloet, 2004).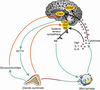 | Figure 13.1 Processus moléculaires mis en jeu par le stress et la dépression (d’après Raison et coll., 2006) |
Une autre hypothèse a émergé, basée sur la théorie « glucocorticoïdes » du vieillissement cérébral (Sapolsky et coll., 1986 ; Landfield et coll., 2007) selon laquelle l’exposition chronique aux glucocorticoïdes serait liée au vieillissement cérébral au niveau de l’hippocampe et à la maladie d’Alzheimer. Selon cette hypothèse, il existerait un continuum entre stress chronique, dépression et maladie d’Alzheimer et le stress agirait à la fois sur le déclenchement et sur la progression de la maladie d’Alzheimer (Sotiropoulos et coll., 2008). À ce stade, les données cliniques et expérimentales restent toutefois peu nombreuses (Anisman et coll., 2008 ; Rothman et Mattson, 2010).
; Landfield et coll., 2007) selon laquelle l’exposition chronique aux glucocorticoïdes serait liée au vieillissement cérébral au niveau de l’hippocampe et à la maladie d’Alzheimer. Selon cette hypothèse, il existerait un continuum entre stress chronique, dépression et maladie d’Alzheimer et le stress agirait à la fois sur le déclenchement et sur la progression de la maladie d’Alzheimer (Sotiropoulos et coll., 2008). À ce stade, les données cliniques et expérimentales restent toutefois peu nombreuses (Anisman et coll., 2008 ; Rothman et Mattson, 2010).La littérature suggère que la résistance aux glucocorticoïdes et l’hyperactivité de l’axe HPA qui en résulte est une cause de la dépression alors que les atrophies de l’hippocampe observées sont une résultante de la dépression, contribuant à des troubles neurocognitifs. Une apparente contradiction existe entre des effets délétères de glucocorticoïdes en excès au niveau de l’hippocampe, médiés par un récepteur GR fonctionnel, alors que ce même excès dans la dépression est le résultat de mécanismes feedback de l’axe HPA déficients, déficience attribuée à un récepteur GR non fonctionnel. Il semble en fait que la fonction GR peut être différente dans différents tissus : un récepteur non fonctionnel peut être observé dans l’hypophyse par exemple, (où il contribue à une déficience des mécanismes de rétrocontrôle et à une hyperactivité de l’axe HPA), alors qu’un récepteur GR fonctionnel sera présent dans l’hippocampe où l’on observera les effets délétères d’un excès de glucocorticoïdes (Anacker et coll., 2010).
).Les mécanismes moléculaires conduisant à l’atrophie neuronale en réponse au stress sont nombreux et restent encore largement à élucider. Ils semblent faire intervenir le glutamate et son récepteur et pourraient conduire à la régulation de la production de facteurs de croissance (BDNF : Brain-Derived Neurotrophic Factor ; vEGF : vascular Endothelial Growth Factor) et à l’activation de nombreux gènes cibles (Pittenger et Duman, 2008).
).Les facteurs de stress psychologiques induisent également la production de cytokines dans le système nerveux central au niveau des cellules microgliales (Dantzer et coll., 2008 ; Miller et coll., 2009). Les cytokines cérébrales pro-inflammatoires (en particulier Il1-b, Il-6 et le TNFa) vont agir sur des cellules neurales et non-neurales par le biais de leurs récepteurs spécifiques et ainsi contribuer au développement de troubles neuropsychiatriques. Les mécanismes par lesquels les cytokines induisent des états dépressifs ont été étudiés en particulier chez des patients traités par immunothérapie (interféron-α par exemple). Ces études montrent que les cytokines influencent le développement de la dépression en agissant sur le métabolisme des monoamines. L’interféron-α ou le TNF-α, par exemple, active une enzyme (indolamine 2,3 dioxygénase) qui métabolise le tryptophane en kynurénine. Or, le tryptophane est aussi le précurseur de la sérotonine qui se trouve ainsi diminuée par l’activation de l’indolamine 2,3 dioxygénase dans le cerveau. Les cytokines auraient également un effet sur la biodisponibilité de la dopamine cérébrale conduisant à un ralentissement psychomoteur et à la fatigue. Enfin, des études d’imagerie cérébrale sur des patients sous immunothérapie montrent l’activation de circuits neuronaux associés à l’anxiété et la vigilance (cortex cingulaire antérieure dorsal).
; Miller et coll., 2009). Les cytokines cérébrales pro-inflammatoires (en particulier Il1-b, Il-6 et le TNFa) vont agir sur des cellules neurales et non-neurales par le biais de leurs récepteurs spécifiques et ainsi contribuer au développement de troubles neuropsychiatriques. Les mécanismes par lesquels les cytokines induisent des états dépressifs ont été étudiés en particulier chez des patients traités par immunothérapie (interféron-α par exemple). Ces études montrent que les cytokines influencent le développement de la dépression en agissant sur le métabolisme des monoamines. L’interféron-α ou le TNF-α, par exemple, active une enzyme (indolamine 2,3 dioxygénase) qui métabolise le tryptophane en kynurénine. Or, le tryptophane est aussi le précurseur de la sérotonine qui se trouve ainsi diminuée par l’activation de l’indolamine 2,3 dioxygénase dans le cerveau. Les cytokines auraient également un effet sur la biodisponibilité de la dopamine cérébrale conduisant à un ralentissement psychomoteur et à la fatigue. Enfin, des études d’imagerie cérébrale sur des patients sous immunothérapie montrent l’activation de circuits neuronaux associés à l’anxiété et la vigilance (cortex cingulaire antérieure dorsal).Devant la multitude des modalités physiopathologiques mises en jeu et le manque de connaissances concernant les distinctions mécanistiques entre les effets du stress aigu et du stress chronique, la question de la relation causale entre stress et dépression reste aujourd’hui largement ouverte (Baune, 2009).
).Concernant les liens entre troubles du sommeil et stress chronique chez l’homme et chez l’animal, une altération du sommeil sur un plan quantitatif et qualitatif est largement décrite : désorganisation des cycles et réduction du temps total de sommeil, fragmentation par éveils multiples, diminution du sommeil lent profond et perturbations de l’organisation du sommeil paradoxal, en particulier raccourcissement de la latence d’apparition du premier épisode de sommeil paradoxal (Mendlewicz et coll., 1991 ; Van Reeth et coll., 2000). Les études impliquant une hypersécrétion de glucocorticoïdes dans ces troubles du sommeil sont largement controversées (Vázquez-Palacios et coll., 2004). La prolactine pourrait être un des médiateurs des effets du stress sur le sommeil (Bodosi et coll., 2000). Le CRF apparaît dans de nombreuses études chez les rongeurs comme le composant clé des effets du stress sur le sommeil, autant de part son rôle au niveau central que dans l’activation de l’axe corticotrope (Chang et Opp, 2001 ; Pawlyk et coll., 2006). Le système sérotoninergique est également un bon candidat : il est largement impliqué chez l’animal dans les effets du stress sur le sommeil et en particulier sur le sommeil paradoxal (Chaouloff et coll., 1999 ; Jouvet, 1999 ; Ursin, 2002).
; Van Reeth et coll., 2000). Les études impliquant une hypersécrétion de glucocorticoïdes dans ces troubles du sommeil sont largement controversées (Vázquez-Palacios et coll., 2004). La prolactine pourrait être un des médiateurs des effets du stress sur le sommeil (Bodosi et coll., 2000). Le CRF apparaît dans de nombreuses études chez les rongeurs comme le composant clé des effets du stress sur le sommeil, autant de part son rôle au niveau central que dans l’activation de l’axe corticotrope (Chang et Opp, 2001 ; Pawlyk et coll., 2006). Le système sérotoninergique est également un bon candidat : il est largement impliqué chez l’animal dans les effets du stress sur le sommeil et en particulier sur le sommeil paradoxal (Chaouloff et coll., 1999 ; Jouvet, 1999 ; Ursin, 2002).On suppose depuis plusieurs années que le stress peut favoriser l’émergence d’une addiction. Dans les situations de stress, de grandes quantités d’hormones de stress, les glucocorticoïdes, sont sécrétées. Or, ces hormones augmentent la sensibilité du cerveau aux psychotropes et favorisent l’émergence de comportements addictifs chez les animaux stressés de manière répétée (Piazza et Le Moal, 1996 et 1998 ; Marinelli et Piazza, 2002). Parallèlement, chez les rats rendus dépendants à une substance, l’administration de molécules qui réduisent l’action des hormones de stress a pour effet de diminuer la consommation des rongeurs (Richardson et coll., 2008 ; Specio et coll., 2008). La sécrétion de glucocorticoïdes est plus ou moins élevée selon les individus et la concentration d’hormone conditionne la susceptibilité à l’addiction (Piazza et Le Moal, 1996). L’inverse a également été récemment vérifié. En effet, des personnes dépendantes à la cocaïne présentent une sensibilité exacerbée aux événements stressants (Fox et coll., 2008). Le stress devient donc un facteur de risque d’une grande importance dans le phénomène de rechute. Des évidences scientifiques existent en faveur d’une convergence des mécanismes d’action des drogues et du stress avec induction de changements similaires au niveau du système dopaminergique mésolimbique (Piazza et Le Moal, 1998) et le rôle central du récepteur des glucocorticoïdes GR dans la médiation de ces effets (De Jong et De Kloet, 2004). Récemment, une équipe de chercheurs français a identifié les neurones impliqués dans la modulation des addictions par le stress : ce sont des neurones sensibles à la fois aux glucocorticoïdes et à la dopamine (Ambroggi et coll., 2009).
et 1998 ; Marinelli et Piazza, 2002). Parallèlement, chez les rats rendus dépendants à une substance, l’administration de molécules qui réduisent l’action des hormones de stress a pour effet de diminuer la consommation des rongeurs (Richardson et coll., 2008 ; Specio et coll., 2008). La sécrétion de glucocorticoïdes est plus ou moins élevée selon les individus et la concentration d’hormone conditionne la susceptibilité à l’addiction (Piazza et Le Moal, 1996). L’inverse a également été récemment vérifié. En effet, des personnes dépendantes à la cocaïne présentent une sensibilité exacerbée aux événements stressants (Fox et coll., 2008). Le stress devient donc un facteur de risque d’une grande importance dans le phénomène de rechute. Des évidences scientifiques existent en faveur d’une convergence des mécanismes d’action des drogues et du stress avec induction de changements similaires au niveau du système dopaminergique mésolimbique (Piazza et Le Moal, 1998) et le rôle central du récepteur des glucocorticoïdes GR dans la médiation de ces effets (De Jong et De Kloet, 2004). Récemment, une équipe de chercheurs français a identifié les neurones impliqués dans la modulation des addictions par le stress : ce sont des neurones sensibles à la fois aux glucocorticoïdes et à la dopamine (Ambroggi et coll., 2009).Maladies métaboliques et affections cardiovasculaires
Les liens entre maladies cardiovasculaires et état de stress chronique (appréhendé principalement par les modèles de job strain de Karasek et ERI (Effort-Reward Imbalance) de Siegrist) ont fait l’objet de nombreuses études épidémiologiques et sont aujourd’hui bien établis (Belkic et coll., 2004). Les résultats épidémiologiques récents montrent également que le stress chronique est associé à une augmentation de l’incidence de l’obésité viscérale et du syndrome métabolique (Chandola et coll., 2006). C’est à nouveau beaucoup moins clair pour les mécanismes physiopathologiques. En cas de stress chronique, les troubles métaboliques occasionnés par la réponse neuroendocrinienne vont agir de concert pour conduire à l’expression clinique d’un certain nombre de comorbidités associant obésité viscérale, hypertension artérielle (HTA), dyslipidémie et dysfonction endothéliale qui sont les composants du syndrome métabolique et font le lit de l’athérosclérose.
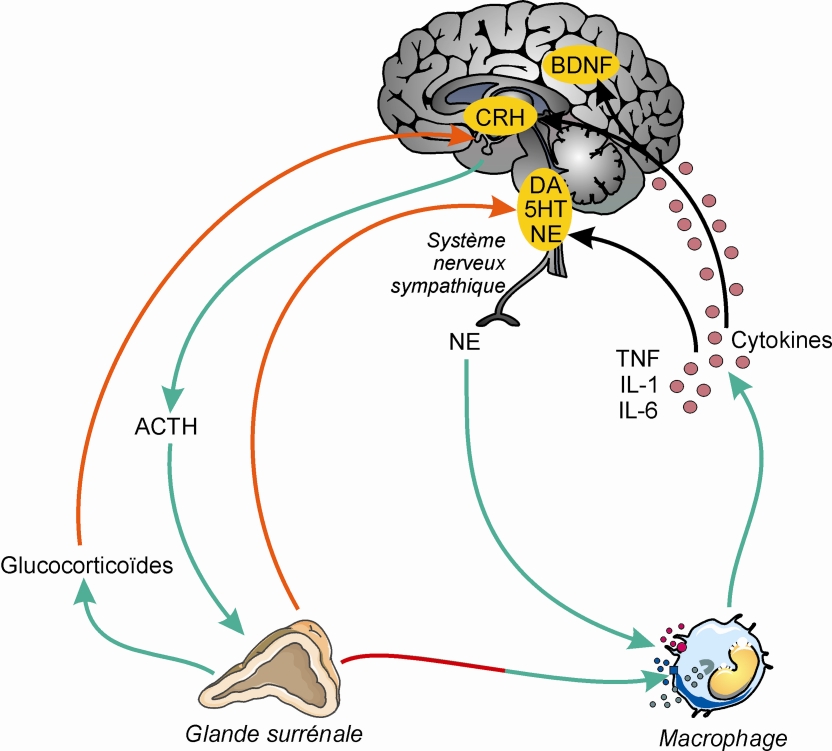
). Les résultats épidémiologiques récents montrent également que le stress chronique est associé à une augmentation de l’incidence de l’obésité viscérale et du syndrome métabolique (Chandola et coll., 2006). C’est à nouveau beaucoup moins clair pour les mécanismes physiopathologiques. En cas de stress chronique, les troubles métaboliques occasionnés par la réponse neuroendocrinienne vont agir de concert pour conduire à l’expression clinique d’un certain nombre de comorbidités associant obésité viscérale, hypertension artérielle (HTA), dyslipidémie et dysfonction endothéliale qui sont les composants du syndrome métabolique et font le lit de l’athérosclérose.Évoquée en 2002 par l’étude transversale de Brunner et coll. (2002), la première évidence scientifique que le stress chronique au travail, via l’hyperactivité de l’axe corticotrope et l’hypersécrétion de catécholamines, pouvait induire un syndrome métabolique a été apportée par les travaux de Chandola et coll. en 2006 (figure 13.2).
, la première évidence scientifique que le stress chronique au travail, via l’hyperactivité de l’axe corticotrope et l’hypersécrétion de catécholamines, pouvait induire un syndrome métabolique a été apportée par les travaux de Chandola et coll. en 2006 (figure 13.2).Le syndrome métabolique est un facteur de risque de diabète de type 2, de maladies cardiovasculaires et d’accidents vasculaires cérébraux (Chandola et coll., 2006). Le cortisol interfère à différents niveaux de la production d’insuline et de l’activation de son récepteur. Le cortisol inhibe directement la sécrétion d’insuline par les cellules β du pancréas. Sur des adipocytes en culture, la déxaméthasone (hormone glucocorticoïde de synthèse) induit progressivement un état de résistance à l’insuline en régulant de multiples aspects des systèmes de transport du glucose (Rosmond, 2005). Le cortisol a également un effet chronique sur le métabolisme des lipides : un excès de cortisol active la lipoprotéine lipase, enzyme qui permet l’hydrolyse des triglycérides des lipoprotéines plasmatiques, aboutissant à une accumulation de triglycérides dans les adipocytes (Rosmond, 2005). Chez la souris, le stress chronique favorise également l’obésité abdominale via le neuropeptide Y (NPY), un neurotransmetteur orexigene libéré directement dans les tissus adipeux (Kuo et coll., 2008). Enfin, le stress chronique provoque une augmentation de la faim avec une appétence marquée pour une nourriture riche en calories, lien supplémentaire avec l’obésité (Teegarden et Bale, 2008). Sous l’effet des glucocorticoïdes, l’humain tend à augmenter sa consommation d’aliments réconfortants (comfort food) et conséquemment son poids corporel (Dallman et coll., 2003).
). Le cortisol interfère à différents niveaux de la production d’insuline et de l’activation de son récepteur. Le cortisol inhibe directement la sécrétion d’insuline par les cellules β du pancréas. Sur des adipocytes en culture, la déxaméthasone (hormone glucocorticoïde de synthèse) induit progressivement un état de résistance à l’insuline en régulant de multiples aspects des systèmes de transport du glucose (Rosmond, 2005). Le cortisol a également un effet chronique sur le métabolisme des lipides : un excès de cortisol active la lipoprotéine lipase, enzyme qui permet l’hydrolyse des triglycérides des lipoprotéines plasmatiques, aboutissant à une accumulation de triglycérides dans les adipocytes (Rosmond, 2005). Chez la souris, le stress chronique favorise également l’obésité abdominale via le neuropeptide Y (NPY), un neurotransmetteur orexigene libéré directement dans les tissus adipeux (Kuo et coll., 2008). Enfin, le stress chronique provoque une augmentation de la faim avec une appétence marquée pour une nourriture riche en calories, lien supplémentaire avec l’obésité (Teegarden et Bale, 2008). Sous l’effet des glucocorticoïdes, l’humain tend à augmenter sa consommation d’aliments réconfortants (comfort food) et conséquemment son poids corporel (Dallman et coll., 2003).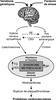 | Figure 13.2 Rôle du stress dans le développement du syndrome métabolique et des pathologies cardiovasculaires (d’après Rosmond, 2005) |

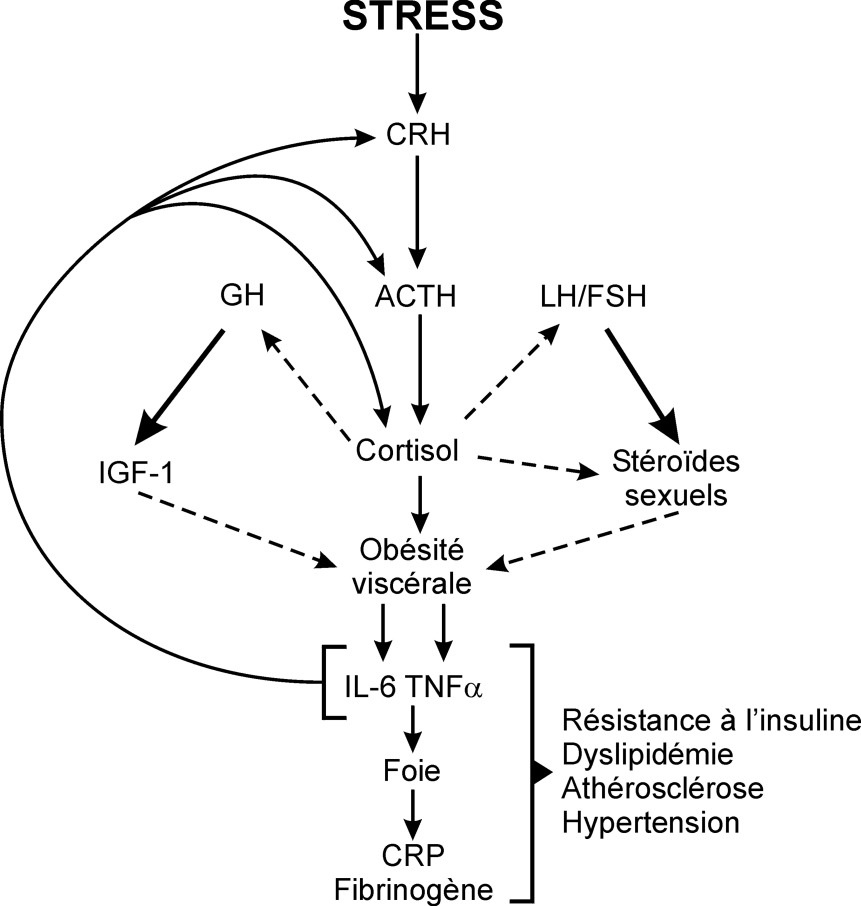
L’hyperactivité de l’axe corticotrope et du système sympathique en cas de stress chronique a une action directe sur l’obésité viscérale : le cortisol supprime l’effet bénéfique des hormones sexuelles et de l’hormone de croissance au niveau viscéral et stimule directement la prolifération des adipocytes (Kyrou et Tsigos, 2007 et 2009). Réciproquement, l’obésité provoque un état inflammatoire médié par les cytokines TNF-α et IL-6, dont la sécrétion est proportionnelle à la masse adipeuse. Ces cytokines pro-inflammatoires sont associées au risque cardiovasculaire et à l’insulinorésistance et stimulent en retour l’axe corticotrope, créant un cercle vicieux délétère (Kyrou et Tsigos, 2007 et 2009) (figure 13.3).
et 2009). Réciproquement, l’obésité provoque un état inflammatoire médié par les cytokines TNF-α et IL-6, dont la sécrétion est proportionnelle à la masse adipeuse. Ces cytokines pro-inflammatoires sont associées au risque cardiovasculaire et à l’insulinorésistance et stimulent en retour l’axe corticotrope, créant un cercle vicieux délétère (Kyrou et Tsigos, 2007 et 2009) (figure 13.3).Le système sympathique est l’autre médiateur majeur du stress sur le système cardiovasculaire. Via les sécrétions d’adrénaline et de noradrénaline, il va agir sur les vaisseaux sanguins (vasoconstriction ou vasodilatation selon les récepteurs), sur le cœur (augmentation de la fréquence cardiaque, de la pression artérielle et du débit cardiaque) et sur le métabolisme (effet lipolytique, effet hyperglycémiant) (Grippo et Johnson, 2009). Les effets du stress sur le système cardiovasculaire peuvent aussi s’expliquer par une action directe sur la variabilité cardiaque (chute de la variabilité cardiaque) via une saturation du système sympathique (et une diminution du système parasympathique) qui aboutit à une instabilité électrique cardiaque (Rosmond, 2005).
). Les effets du stress sur le système cardiovasculaire peuvent aussi s’expliquer par une action directe sur la variabilité cardiaque (chute de la variabilité cardiaque) via une saturation du système sympathique (et une diminution du système parasympathique) qui aboutit à une instabilité électrique cardiaque (Rosmond, 2005).Enfin, les liens entre stress chronique et altération de la structure et de la quantité de sommeil (Van Reeth et coll., 2000) ouvrent la voie à d’autres mécanismes potentiels via la dette de sommeil comme facteur de risque de maladie métabolique (Spiegel et coll., 1999 ; Knutson et coll., 2007). On sait aujourd’hui qu’une dette de sommeil est susceptible d’augmenter la sévérité de maladies comme l’obésité, le diabète, l’hypertension et la vulnérabilité aux infections (Ohlmann et O’Sullivan, 2009). Ces dernières années, plusieurs études épidémiologiques ont établi le lien entre un sommeil court et un indice de masse corporelle (IMC) élevé, à la fois chez l’adulte et l’enfant (Nielsen et coll., 2010). Deux hormones clés sont impliquées dans la régulation du comportement alimentaire : la ghréline, sécrétée par l’estomac et qui stimule l’appétit ; la leptine, produite par les cellules adipeuses et qui induit la satiété. Spiegel et coll. (2009) ont montré qu’une réduction de la durée de sommeil était associée à une diminution de la leptine anorexigène et à une augmentation de la ghréline orexigène, et que ces modifications hormonales étaient effectivement associées à une augmentation de faim et d’appétit en particulier pour les aliments riches en graisses et en sucre (Spiegel et coll., 2009).
) ouvrent la voie à d’autres mécanismes potentiels via la dette de sommeil comme facteur de risque de maladie métabolique (Spiegel et coll., 1999 ; Knutson et coll., 2007). On sait aujourd’hui qu’une dette de sommeil est susceptible d’augmenter la sévérité de maladies comme l’obésité, le diabète, l’hypertension et la vulnérabilité aux infections (Ohlmann et O’Sullivan, 2009). Ces dernières années, plusieurs études épidémiologiques ont établi le lien entre un sommeil court et un indice de masse corporelle (IMC) élevé, à la fois chez l’adulte et l’enfant (Nielsen et coll., 2010). Deux hormones clés sont impliquées dans la régulation du comportement alimentaire : la ghréline, sécrétée par l’estomac et qui stimule l’appétit ; la leptine, produite par les cellules adipeuses et qui induit la satiété. Spiegel et coll. (2009) ont montré qu’une réduction de la durée de sommeil était associée à une diminution de la leptine anorexigène et à une augmentation de la ghréline orexigène, et que ces modifications hormonales étaient effectivement associées à une augmentation de faim et d’appétit en particulier pour les aliments riches en graisses et en sucre (Spiegel et coll., 2009).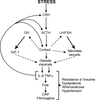 | Figure 13.3 Représentation schématique des relations réciproques entre le stress chronique et l’obésité viscérale, aboutissant au syndrome métabolique (d’après Kyrou et Tsigos, 2008) |
Pathologies digestives
Des événements de vie dits stressants sont souvent évoqués par les malades comme facteurs responsables du déclenchement et/ou de la majoration de leurs symptômes digestifs. Une meilleure connaissance des mécanismes impliqués dans la réponse au stress a permis de mieux appréhender l’imputabilité du stress dans les domaines essentiels de la pathologie digestive que sont les troubles fonctionnels digestifs (TFD) et les maladies inflammatoires cryptogénétiques de l’intestin (MICI) : maladie de Crohn (MC) et rectocolite hémorragique (RCH). Toutefois, nos connaissances concernent essentiellement les stress aigus, alors que celles concernant des stress chroniques, plus proches des préoccupations cliniques et du stress au travail, demeurent très fragmentaires.
Les TFD constituent un ensemble de syndromes classiquement dissocié en dyspepsie non ulcéreuse, douleur thoracique non angineuse, troubles fonctionnels intestinaux encore dénommés « syndrome de l’intestin irritable » (SII) correspondant à l’ancienne appellation de colopathie fonctionnelle. Le rôle aggravant du stress sur les symptômes est fréquemment souligné par les malades souffrant d’une dyspepsie fonctionnelle et surtout d’un SII. Les événements de vie douloureux (séparation, deuil, situation financière critique, chômage...), lorsqu’ils sont perçus comme une menace, sont particulièrement associés à l’exacerbation des symptômes, souvent de façon transitoire. Le stress pourrait également jouer un rôle dans l’apparition des symptômes car environ un malade sur deux rapporte une relation chronologique entre la survenue d’un stress et l’apparition des premiers symptômes de TFD (Taché et coll., 2008).
).L’expérimentation animale a permis d’approcher les mécanismes des effets moteurs gastriques et coliques au cours d’un stress aigu. Ils impliquent l’action centrale du CRF agissant par l’intermédiaire de deux types de récepteurs. L’effet moteur inhibiteur gastrique du CRF implique le récepteur de type 2 (CRF2) alors que l’effet stimulant sur la motricité colique met en jeu les récepteurs de type 1 (CRF1) (Taché et Bonaz, 2007 ; Taché et coll., 2008).
; Taché et coll., 2008).L’étiopathogénie des MICI (MC et RCH) est multifactorielle impliquant des facteurs immunologiques, génétiques, infectieux ou environnementaux. Des travaux récents ont apporté de solides arguments en faveur de l’existence d’une relation entre stress et évolution des MICI. Parallèlement, un effet pro-inflammatoire du stress au niveau du tube digestif a été démontré, impliquant les lymphocytes T CD4+ (Maunder et coll., 2008). Le stress pourrait non seulement jouer un rôle dans le déclenchement d’une poussée de MICI mais également dans l’apparition de la maladie. Sur le plan physiopathogénique, Gué et coll. (1997) ont montré chez le rat que l’aggravation d’une colite aiguë par un stress n’impliquait pas le CRF et l’arginine vasopressine (AVP). En fait, selon d’autres travaux, le CRF central empêcherait l’aggravation de l’inflammation colique. En effet, les rats Lewis femelles connus pour avoir un défaut de sécrétion de CRF sont plus sensibles aux infections ou aux inflammations. À l’opposé, les rats Fischer, qui ont une hypersécrétion de CRF, sont plus résistants aux infections et/ou aux inflammations (Stöhr et coll., 2000). Le CRF renforcerait l’immunité humorale aux dépens de l’immunité cellulaire, en stimulant préférentiellement la production de cytokines immuno-régulatrices de type 2 (TH-2 pour T-helper 2), IL-4 et IL-5 par rapport à celles de type 1 (TH-1 pour T-helper 1), IFN g et IL-2. Alors que le CRF central a un rôle protecteur de l’inflammation, le CRF périphérique a plutôt un rôle pro-inflammatoire (Dhabhar, 2009).
). Le stress pourrait non seulement jouer un rôle dans le déclenchement d’une poussée de MICI mais également dans l’apparition de la maladie. Sur le plan physiopathogénique, Gué et coll. (1997) ont montré chez le rat que l’aggravation d’une colite aiguë par un stress n’impliquait pas le CRF et l’arginine vasopressine (AVP). En fait, selon d’autres travaux, le CRF central empêcherait l’aggravation de l’inflammation colique. En effet, les rats Lewis femelles connus pour avoir un défaut de sécrétion de CRF sont plus sensibles aux infections ou aux inflammations. À l’opposé, les rats Fischer, qui ont une hypersécrétion de CRF, sont plus résistants aux infections et/ou aux inflammations (Stöhr et coll., 2000). Le CRF renforcerait l’immunité humorale aux dépens de l’immunité cellulaire, en stimulant préférentiellement la production de cytokines immuno-régulatrices de type 2 (TH-2 pour T-helper 2), IL-4 et IL-5 par rapport à celles de type 1 (TH-1 pour T-helper 1), IFN g et IL-2. Alors que le CRF central a un rôle protecteur de l’inflammation, le CRF périphérique a plutôt un rôle pro-inflammatoire (Dhabhar, 2009).Parallèlement aux systèmes CRF central et périphérique, le système nerveux autonome est très impliqué dans les relations stress-inflammation digestive (Vere et coll., 2009). Il existe classiquement, au cours d’un stress aigu, une activation du système sympathique et une inhibition du système parasympathique. Or, le système sympathique a un rôle délétère sur l’inflammation. En activant le système sympathique, le stress altérerait les fonctions immunitaires, augmenterait la perméabilité intestinale et favoriserait des modifications du mucus. La noradrénaline en tant que neurotransmetteur ou les catécholamines circulantes affectent la circulation et la prolifération lymphocytaire et modulent la production de cytokines et l’activité fonctionnelle de diverses cellules lymphoïdes. Le stress entraîne aussi une augmentation de la perméabilité intestinale, une augmentation de la motilité intestinale et altère la sécrétion ionique (Caso et coll., 2008). L’effet sur la barrière intestinale serait un des éléments à l’origine de poussées de MICI et ferait intervenir le mastocyte dont le rôle est central dans les phénomènes de perméabilité intestinale (Keita et coll., 2010).
). Il existe classiquement, au cours d’un stress aigu, une activation du système sympathique et une inhibition du système parasympathique. Or, le système sympathique a un rôle délétère sur l’inflammation. En activant le système sympathique, le stress altérerait les fonctions immunitaires, augmenterait la perméabilité intestinale et favoriserait des modifications du mucus. La noradrénaline en tant que neurotransmetteur ou les catécholamines circulantes affectent la circulation et la prolifération lymphocytaire et modulent la production de cytokines et l’activité fonctionnelle de diverses cellules lymphoïdes. Le stress entraîne aussi une augmentation de la perméabilité intestinale, une augmentation de la motilité intestinale et altère la sécrétion ionique (Caso et coll., 2008). L’effet sur la barrière intestinale serait un des éléments à l’origine de poussées de MICI et ferait intervenir le mastocyte dont le rôle est central dans les phénomènes de perméabilité intestinale (Keita et coll., 2010).Concernant les pathologies ulcéreuses de l’estomac, une infection par Helicobacter pylori est généralement associée à la survenue d’un ulcère. Toutefois, quelques données cliniques ont suggéré que cette infection n’était pas suffisante en soi pour provoquer un ulcère : des patients infectés par la bactérie ne développent pas d’ulcère et des patients atteints d’ulcère gastrique ont une sérologie H-pylori négative (Velin et Michetti, 2006). Chez la souris, il a été montré récemment que l’infection de l’estomac par Helicobacter pylori est potentialisée par un stress psychologique (souris mises en présence de congénères subissant des chocs électriques), et que les glucocorticoïdes sont responsables de cet effet (Guo et coll., 2009). En fait, la revue de la littérature chez l’homme suggère que d’autres facteurs tels la consommation de médicaments anti-inflammatoires non stéroïdiens, le régime alimentaire, le tabagisme et le stress pourraient contribuer au développement d’un ulcère peptique (gastro-duodénal) (Kurata et Nowaga, 1997), mais les cas d’ulcères sans infection d’Helicobacter pylori restent très marginaux (revue dans Gisbert et Calvet, 2009).
). Chez la souris, il a été montré récemment que l’infection de l’estomac par Helicobacter pylori est potentialisée par un stress psychologique (souris mises en présence de congénères subissant des chocs électriques), et que les glucocorticoïdes sont responsables de cet effet (Guo et coll., 2009). En fait, la revue de la littérature chez l’homme suggère que d’autres facteurs tels la consommation de médicaments anti-inflammatoires non stéroïdiens, le régime alimentaire, le tabagisme et le stress pourraient contribuer au développement d’un ulcère peptique (gastro-duodénal) (Kurata et Nowaga, 1997), mais les cas d’ulcères sans infection d’Helicobacter pylori restent très marginaux (revue dans Gisbert et Calvet, 2009).Le stress psychosocial influence également l’évolution des hépatites virales, cirrhoses et carcinomes hépatiques (Vere et coll., 2009). Le stress influencerait l’évolution de l’inflammation hépatique en augmentant le niveau des cytokines pro-inflammatoires IL-6 et TNFa. Le stress semble également influencer la progression tumorale en agissant, par l’intermédiaire de cytokines, sur l’activité des cellules NKT (Natural Killer T) (Vere et coll., 2009). Des modèles animaux sont en cours d’élaboration pour affiner ces mécanismes.
). Le stress influencerait l’évolution de l’inflammation hépatique en augmentant le niveau des cytokines pro-inflammatoires IL-6 et TNFa. Le stress semble également influencer la progression tumorale en agissant, par l’intermédiaire de cytokines, sur l’activité des cellules NKT (Natural Killer T) (Vere et coll., 2009). Des modèles animaux sont en cours d’élaboration pour affiner ces mécanismes.Troubles musculosquelettiques (TMS)
Les TMS, ou troubles musculosquelettiques, sont des maladies multifactorielles à composante professionnelle. Les sollicitations qui sont à l’origine des TMS sont biomécaniques, organisationnelles et psychosociales. Les TMS, dont certains sont reconnus comme maladies professionnelles (tableaux 57, 69, 79, 97, 98)2
, peuvent concerner les membres supérieurs, le dos et les membres inférieurs. Les plus fréquents sont le syndrome du canal carpien, les tendinites (épaules, coudes...) et les atteintes du rachis (lombalgies...).
L’existence d’un lien entre troubles musculosquelettiques et stress est largement suspectée suite à des études épidémiologiques qui ont montré une association entre les deux (revue dans Macfarlane et coll., 2009). La question d’une relation entre stress et TMS est posée tant du point de vue des phénomènes de causalité : « quoi favorise quoi », que du point vue de plausibilité biologique : « comment est-ce possible ? ». Dans un article de synthèse, Aptel et Cnockaert (2002) évoquent 4 voies mécanistiques possibles (figure 13.4).
). La question d’une relation entre stress et TMS est posée tant du point de vue des phénomènes de causalité : « quoi favorise quoi », que du point vue de plausibilité biologique : « comment est-ce possible ? ». Dans un article de synthèse, Aptel et Cnockaert (2002) évoquent 4 voies mécanistiques possibles (figure 13.4).
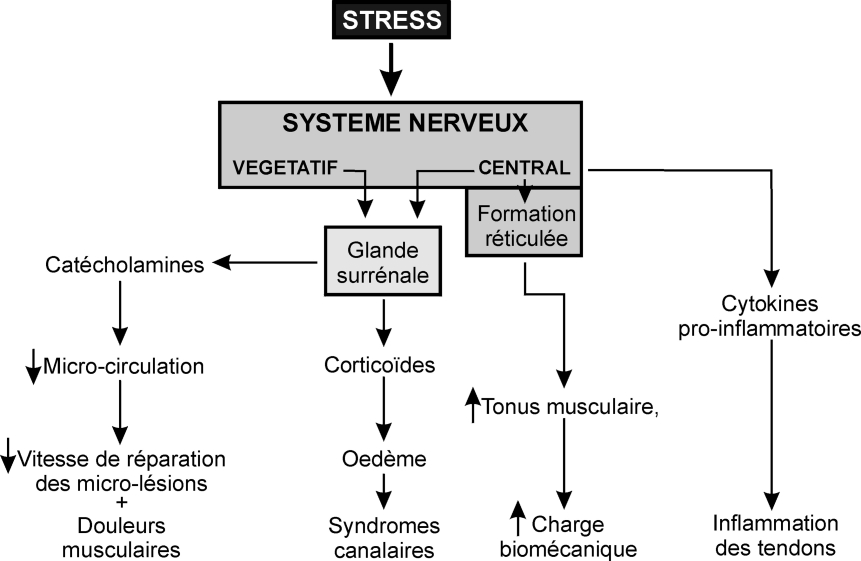
Premièrement, le stress via l’activation du système nerveux central accroît le niveau d’activité (« tonus ») de la formation réticulée localisée dans le tronc cérébral, laquelle à son tour augmente le tonus musculaire. Cette augmentation de tonus musculaire accroît la « charge biomécanique » des muscles et des tendons et contribue ainsi à augmenter le risque de TMS (Aptel et Cnockaert, 2002 ; Nilsen et coll., 2007).
; Nilsen et coll., 2007).Une deuxième voie sollicite le système nerveux végétatif qui déclenche la sécrétion des catécholamines (adrénaline et noradrénaline). Ces substances libérées dans le sang provoquent, entre autres, une augmentation du tonus réticulaire, de la fréquence cardiaque et une vasoconstriction des artérioles. Pour ce qui concerne les TMS, la restriction de la micro-circulation dans le muscle et au voisinage des tendons, dont la vascularisation est par ailleurs pauvre, a deux types d’effets : d’une part, elle réduit l’apport de nutriments aux tendons et ainsi entrave les processus d’auto-réparation des micro-lésions des fibres tendineuses consécutives aux contraintes biomécaniques excessives et d’autre part, elle favorise l’apparition de la fatigue musculaire chronique et de myalgies (Aptel et Cnockaert, 2002 ; Discher et coll., 2009).
; Discher et coll., 2009).Une autre voie explorée est celle conduisant à la libération de glucocorticoïdes par la glande corticosurrénale. Corticostérone et cortisol agissent sur le rein et peuvent perturber l’équilibre hydrominéral de l’organisme dont la conséquence la plus visible est l’œdème. Pour ce qui concerne les TMS, l’œdème peut déclencher des « syndromes canalaires » résultant de la compression locale des nerfs par les tissus adjacents (tendons...) œdématiés (Aptel et Cnockaert, 2002).
).Enfin, le stress pourrait également agir sur le système immunitaire via la production/libération de cytokines. Certaines de ces cytokines, telles les interleukines (IL-1, IL-2, IL-10...) sont pro-inflammatoires. Pour ce qui concerne les TMS, ces interleukines favoriseraient voire provoqueraient des TMS (inflammation des tendons). Cette dernière hypothèse a été confirmée indirectement par les résultats d’une étude sur les effets secondaires d’une trithérapie cancéreuse qui associait deux médicaments spécifiques et l’IL-2. Les patients ainsi traités ont été victimes d’un syndrome du canal carpien trois semaines seulement après le début du traitement. Des essais croisés chez des patients au repos complet, dont les poignets ne subissaient donc pas de contraintes biomécaniques particulières, ont confirmé que l’IL-2 était bien la seule responsable du syndrome du canal carpien (Puduvalli et coll., 1996).
).L’ensemble de ces hypothèses mécanistiques trouvent leur place, de façon très détaillée, dans un modèle novateur de compréhension, le modèle intégré de Bruxelles (Johansson et coll., 2003). Pour reprendre une terminologie de Michel Aptel, le modèle de Bruxelles est le « chaînon manquant » entre les facteurs psychosociaux et la physiopathologie des TMS (Aptel, 2007). Il s’agit d’un modèle holistique intégrant l’ensemble des mécanismes connus pouvant conduire à des myalgies liées au travail. Selon les auteurs (Johansson et coll., 2003), trois dimensions caractérisent le modèle : la diversité de nature des facteurs de risque psychosociaux et biomécaniques et partant, des mécanismes physiopathologiques qui les portent ; l’interaction étroite de ces mécanismes et de leurs rétrocontrôles qui témoigne de l’absence d’un mécanisme dominant ; la dimension chrono-dépendante du modèle qui induit des types différents d’expressions du processus physiopathologique en raison de la multiplicité des voies d’actions en jeu.
). Pour reprendre une terminologie de Michel Aptel, le modèle de Bruxelles est le « chaînon manquant » entre les facteurs psychosociaux et la physiopathologie des TMS (Aptel, 2007). Il s’agit d’un modèle holistique intégrant l’ensemble des mécanismes connus pouvant conduire à des myalgies liées au travail. Selon les auteurs (Johansson et coll., 2003), trois dimensions caractérisent le modèle : la diversité de nature des facteurs de risque psychosociaux et biomécaniques et partant, des mécanismes physiopathologiques qui les portent ; l’interaction étroite de ces mécanismes et de leurs rétrocontrôles qui témoigne de l’absence d’un mécanisme dominant ; la dimension chrono-dépendante du modèle qui induit des types différents d’expressions du processus physiopathologique en raison de la multiplicité des voies d’actions en jeu.Cancer
La littérature épidémiologique liant stress et cancer n’est pas aussi conséquente que pour d’autres pathologies. En juin 2002, le British Medical Journal s’élevait contre cette croyance qui pouvait conduire les personnes atteintes d’un cancer à se culpabiliser. Le journal soulignait notamment qu’il n’existait pas de preuve sérieuse permettant d’affirmer que le stress puisse être la cause du cancer du sein (Graham et coll., 2002). Aujourd’hui, il existe des études suggérant que les changements physiologiques associés au stress chronique pourraient jouer un rôle dans le déclenchement et la progression des tumeurs (Kiecolt-Glaser et Glaser, 1999 ; Chida et coll., 2008). Les arguments scientifiques en faveur d’un rôle du stress dans l’étiologie des cancers sont cependant controversés. De nombreux biais méthodologiques sont relevés dans les études épidémiologiques, expliquant des résultats souvent inconsistants voire contradictoires (Reiche et coll., 2004 ; Schraub et coll., 2009 ; Lopez et coll., 2010).
). Aujourd’hui, il existe des études suggérant que les changements physiologiques associés au stress chronique pourraient jouer un rôle dans le déclenchement et la progression des tumeurs (Kiecolt-Glaser et Glaser, 1999 ; Chida et coll., 2008). Les arguments scientifiques en faveur d’un rôle du stress dans l’étiologie des cancers sont cependant controversés. De nombreux biais méthodologiques sont relevés dans les études épidémiologiques, expliquant des résultats souvent inconsistants voire contradictoires (Reiche et coll., 2004 ; Schraub et coll., 2009 ; Lopez et coll., 2010).Si le rôle des événements de vie, tels que le deuil, et plus généralement du stress est souvent invoqué par la croyance populaire comme cause du cancer, les arguments scientifiques sont aujourd’hui insuffisants. Des études expérimentales et cliniques montrent que le stress et l’isolation sociale chronique contribuent à la progression de certains cancers, en particulier le cancer du sein chez la femme (Fox et coll., 1994 ; Lillberg et coll., 2003). Une dérégulation de l’axe corticotrope, en particulier une altération de rythme circadien de cortisol, semble liée à la mortalité par cancer du sein. Le stress pourrait agir directement via des altérations hormonales ou indirectement via une altération de l’efficacité des traitements anticancéreux (timing d’administration) due à la modification des rythmes circadiens par le stress (Sephton et coll., 2000 ; Sephton et Spiegel, 2003). Chez les rats de la souche Sprague-Dawley, un bon modèle d’étude du cancer du sein, l’amplitude de la sécrétion de corticostérone en réponse au stress ainsi que le temps de récupération de la réponse prédisent la vitesse de croissance des tumeurs (Yee, 2008). L’implication du système CRF (neuropeptides et récepteurs) dans la régulation du développement de certains cancers chez l’homme est également largement suspectée et ouvre la voie à de nouvelles thérapeutiques prometteuses via des agonistes et antagonistes des récepteurs au CRF (Wang et Li, 2007 ; pour revue, Kaprara et coll., 2010).
; Lillberg et coll., 2003). Une dérégulation de l’axe corticotrope, en particulier une altération de rythme circadien de cortisol, semble liée à la mortalité par cancer du sein. Le stress pourrait agir directement via des altérations hormonales ou indirectement via une altération de l’efficacité des traitements anticancéreux (timing d’administration) due à la modification des rythmes circadiens par le stress (Sephton et coll., 2000 ; Sephton et Spiegel, 2003). Chez les rats de la souche Sprague-Dawley, un bon modèle d’étude du cancer du sein, l’amplitude de la sécrétion de corticostérone en réponse au stress ainsi que le temps de récupération de la réponse prédisent la vitesse de croissance des tumeurs (Yee, 2008). L’implication du système CRF (neuropeptides et récepteurs) dans la régulation du développement de certains cancers chez l’homme est également largement suspectée et ouvre la voie à de nouvelles thérapeutiques prometteuses via des agonistes et antagonistes des récepteurs au CRF (Wang et Li, 2007 ; pour revue, Kaprara et coll., 2010).Une autre voie de recherche s’intéressant aux connexions possibles entre stress et cancer concerne la réactivation de virus latents qui favoriseraient le développement de tumeurs (exemples du virus d’Epstein Barr et du lymphome non hodgkinien) (Godbout et Glaser, 2006). Le stress, de par le shift qu’il engendre au niveau du profil des cytokines (d’un type TH-1 vers un type TH-2), pourrait favoriser la réplication de virus et augmenter la fréquence des tumeurs (Glaser et coll., 2001). En résumé, ces études avancent l’hypothèse que le stress via une dérégulation du système immunitaire pourrait être un cofacteur dans la promotion de certaines tumeurs, en particulier celles induites par des virus oncogènes.
). Le stress, de par le shift qu’il engendre au niveau du profil des cytokines (d’un type TH-1 vers un type TH-2), pourrait favoriser la réplication de virus et augmenter la fréquence des tumeurs (Glaser et coll., 2001). En résumé, ces études avancent l’hypothèse que le stress via une dérégulation du système immunitaire pourrait être un cofacteur dans la promotion de certaines tumeurs, en particulier celles induites par des virus oncogènes.Le stress est également associé à une altération de l’immunité anti-tumorale via une réduction de l’activité des cellules Natural Killer (NK) et des cellules T cytotoxiques, cellules normalement impliquées dans la destruction de cellules anormales (Antoni et coll., 2006 ; pour revue, Webster Marketon et Glaser, 2008). Une étude récente chez la souris suggère que des hormones thyroïdiennes pourraient être impliquées dans la médiation des effets du stress sur l’immunité (cellules T) et le cancer (Frick et coll., 2009).
; pour revue, Webster Marketon et Glaser, 2008). Une étude récente chez la souris suggère que des hormones thyroïdiennes pourraient être impliquées dans la médiation des effets du stress sur l’immunité (cellules T) et le cancer (Frick et coll., 2009).Immunité
Les stresseurs psychosociaux affectent la circulation et l’activité des cellules du système immunitaire à travers la libération de médiateurs neuroendocriniens et via des actions neurales directes du système sympathique, parasympathique et peptidergique. Les voies neuronales majeures, à partir desquelles le stress peut affecter les fonctions immunitaires périphériques, sont l’axe néocortico-sympathique, l’axe hypothalamo-hypophyso-surrénalien et la voie tronc cérébral - nerf vague - acétylcholine induisant la libération des médiateurs majeurs : noradrénaline, cortisol et acétylcholine. Ces hormones et neurotransmetteurs peuvent moduler les processus inflammatoires dans les maladies auto-immunes telles que l’arthrite rhumatoïde, la sclérose en plaque ou les pathologies de la peau, ainsi qu’affecter la réponse immunitaire lors d’infection et influencer le développement et la progression de tumeur (Kemeny et Schedlowski, 2007). Les organes lymphoïdes primaires et secondaires sont innervés par des fibres nerveuses noradrénergiques. Toutes les cellules lymphoïdes expriment des adrénorécepteurs β et une partie d’entre elles des adrénorécepteurs α. L’adrénaline et la noradrénaline peuvent ainsi altérer la circulation de sous-populations de leucocytes ainsi que la capacité fonctionnelle de cellules immuno-compétentes incluant la production et la libération de cytokines. Les glucocorticoïdes régulent de multiples aspects des fonctions immunitaires avec des effets anti-inflammatoires et immunosuppresseurs. Ces hormones régulent la réponse immunitaire innée aux infections bactériennes et virales en induisant un glissement de l’activité cellulaire de type T-H1 vers T- H2 par inhibition de la production de cytokines pro-inflammatoires et la stimulation de la synthèse de protéines anti-inflammatoires. Récemment, la voie anti-inflammatoire cholinergique a été mise en évidence. On parle de réflexe inflammatoire comprenant un bras afférent qui détecte via le nerf vague l’inflammation et un bras efférent qui inhibe les réponses immunes innées (Rosas-Ballina et Tracey, 2009).
). Les organes lymphoïdes primaires et secondaires sont innervés par des fibres nerveuses noradrénergiques. Toutes les cellules lymphoïdes expriment des adrénorécepteurs β et une partie d’entre elles des adrénorécepteurs α. L’adrénaline et la noradrénaline peuvent ainsi altérer la circulation de sous-populations de leucocytes ainsi que la capacité fonctionnelle de cellules immuno-compétentes incluant la production et la libération de cytokines. Les glucocorticoïdes régulent de multiples aspects des fonctions immunitaires avec des effets anti-inflammatoires et immunosuppresseurs. Ces hormones régulent la réponse immunitaire innée aux infections bactériennes et virales en induisant un glissement de l’activité cellulaire de type T-H1 vers T- H2 par inhibition de la production de cytokines pro-inflammatoires et la stimulation de la synthèse de protéines anti-inflammatoires. Récemment, la voie anti-inflammatoire cholinergique a été mise en évidence. On parle de réflexe inflammatoire comprenant un bras afférent qui détecte via le nerf vague l’inflammation et un bras efférent qui inhibe les réponses immunes innées (Rosas-Ballina et Tracey, 2009).En condition d’inflammation chronique telle que l’arthrite rhumatoïde, les études expérimentales conduites chez l’homme ont démontré que les communications entre les systèmes neuroendocriniens et immunitaires sont perturbées lors d’une exposition à un stresseur et que ces mécanismes sont à la base de l’aggravation par le stress de ces pathologies. Les données montrent une sécrétion inadéquate de cortisol accompagnée d’une augmentation du tonus sympathique au repos. Lors d’un stress, la réponse de l’axe hypothalamo-hypophyso-surrénalien et du système sympathique est altérée chez ces patients avec une perte fonctionnelle des fibres nerveuses sympathiques synoviales. Ces modifications s’accompagnent d’un déplacement de l’expression des adrénorécepteurs β vers les adrénorécepteurs α et d’une perturbation de la cascade de signalisation intra-cellulaire des adrénorécepteurs au niveau des leucocytes.
De même, chez les patients souffrant de sclérose en plaque, des événements de vie stressants sont liés à une exacerbation des symptômes d’après une méta-analyse de 14 études (Mohr et coll., 2004). Les facteurs de stress ne seraient pas directement la cause des exacerbations de la sclérose en plaque mais ils rendraient les patients vulnérables aux processus d’auto-réactivité immunitaires de la sclérose en plaque conduisant à terme aux exacerbations. En effet, en situation de stress chronique, une résistance aux hormones glucocorticoïdes se développe avec pour conséquence majeure la perte du pouvoir anti-inflammatoire de ces hormones. Dans ces conditions, l’inflammation auto-réactive reste incontrôlée et se poursuit jusqu’à ce que l’exacerbation éclate (Mohr, 2007). Il est important de souligner que des études se développent sur le rôle des facteurs psychologiques et sociaux dans l’interaction entre le stress et la sclérose en plaque. Les capacités d’ajustement (coping style) de l’individu et la présence de support social semblent modérer l’impact du stress sur l’exacerbation de la sclérose en plaque.
). Les facteurs de stress ne seraient pas directement la cause des exacerbations de la sclérose en plaque mais ils rendraient les patients vulnérables aux processus d’auto-réactivité immunitaires de la sclérose en plaque conduisant à terme aux exacerbations. En effet, en situation de stress chronique, une résistance aux hormones glucocorticoïdes se développe avec pour conséquence majeure la perte du pouvoir anti-inflammatoire de ces hormones. Dans ces conditions, l’inflammation auto-réactive reste incontrôlée et se poursuit jusqu’à ce que l’exacerbation éclate (Mohr, 2007). Il est important de souligner que des études se développent sur le rôle des facteurs psychologiques et sociaux dans l’interaction entre le stress et la sclérose en plaque. Les capacités d’ajustement (coping style) de l’individu et la présence de support social semblent modérer l’impact du stress sur l’exacerbation de la sclérose en plaque.Les patients atteints de psoriasis ou de dermatite atopique diffèrent également dans leur réponse à un stresseur psychologique. Les facteurs de stress déclenchent ou aggravent l’inflammation de la peau par des mécanismes encore mal connus mais faisant intervenir l’activité des mastocytes, des cellules « Natural Killer » ou des cellules dendritiques de la peau. Cette activité est régulée par les médiateurs de l’axe hypothalamo-hypophyso-surrénalien (CRH, ACTH pour Adrenocorticotropin Hormone, et glucocorticoïdes) ainsi que par les catécholamines et la substance P.
Les relations entre les facteurs de stress et les maladies infectieuses sont très étudiées, en particulier les challenges viraux, la réponse aux vaccinations et la réactivation de virus latents. Le challenge viral consiste à inoculer des individus sains avec un virus dans des conditions contrôlées. Des événements de vie stressants, le stress perçu et les émotions négatives prédisent une plus grande susceptibilité aux infections rhinovirales et au virus influenza, des titres d’anticorps plus bas et une sécrétion plus élevée de l’interleukine-6. Les mécanismes impliqués dans l’effet du stress sur la résistance au virus influenza proviennent d’études chez l’animal et proposent que les voies principales mettent en jeu la réponse des cytokines pro-inflammatoires, les chémokines β, et les cellules « Natural Killer ». La mesure de l’immunité suite à une vaccination contre le virus de l’influenza ou l’hépatite B montre une variabilité individuelle qui est influencée par des facteurs de stress tels qu’une période d’examen chez des étudiants ou le stress chronique induit par l’attention apportée à un proche souffrant de la maladie d’Alzheimer. Enfin, la réactivation de virus latents tels que le virus d’Epstein-Barr, le virus de l’herpès HSV-1 ou le cytomégalovirus a été étudiée dans des conditions stressantes. Le stress induit par des examens académiques par exemple est capable de réactiver ce type de virus latents et d’augmenter le titre des anticorps (Glaser, 2005).
).Plusieurs études ont montré l’influence du stress sur la progression du virus VIH par la perte plus rapide de cellules T CD4. Des études chez le macaque ont appuyé ces données montrant que des stresseurs sociaux telles que des séparations ou des changements de logement de macaques inoculés avec le virus de l’immunodéficience simienne (VIS) accéléraient la progression de la maladie et les altérations immunes associées. La noradrénaline, plutôt que le cortisol, semble impliquée dans ce processus en stimulant la réplication du virus VIH (Miller et coll., 2009). Des interventions psychologiques de gestion du stress chez des patients séropositifs ont résulté par exemple, en une réduction des titres d’anticorps contre des virus de l’herpès (EBV pour Epstein-Barr Virus, HSV-2 pour Herpes Simplex Virus 2) ainsi que des humeurs dépressives (Carrico et Antoni, 2008).
). Des interventions psychologiques de gestion du stress chez des patients séropositifs ont résulté par exemple, en une réduction des titres d’anticorps contre des virus de l’herpès (EBV pour Epstein-Barr Virus, HSV-2 pour Herpes Simplex Virus 2) ainsi que des humeurs dépressives (Carrico et Antoni, 2008).En plus d’une libération périphérique, les facteurs de stress psychologiques induisent la production de cytokines dans le système nerveux central au niveau des cellules microgliales (Dantzer et coll., 2008 ; Miller, 2008), contribuant au développement de troubles neuropsychiatriques, comme indiqué dans la partie « santé mentale » de ce chapitre.
; Miller, 2008), contribuant au développement de troubles neuropsychiatriques, comme indiqué dans la partie « santé mentale » de ce chapitre.
En conclusion, l’altération de la régulation de l’axe corticotrope en cas de stress chronique apparaît impliquée dans les troubles de l’humeur : des augmentations de sécrétions basales de cortisol ont souvent été rapportées chez l’homme. Des anomalies circadiennes (avances de phase du rythme de cortisol) sont suspectées comme lien possible entre stress chronique et dépression. L’axe corticotrope intervient également largement dans la modulation des comportements addictifs par le stress et dans les troubles du sommeil liés au stress. L’hypersécrétion de cortisol et de catécholamines en cas de stress chronique peut conduire à l’apparition d’un syndrome métabolique associant plusieurs symptômes : obésité abdominale, état de résistance à l’insuline pouvant évoluer vers un diabète, hypertension artérielle et perturbations du métabolisme des lipides sanguins. Ces perturbations métaboliques représentent un facteur de risque pour le système cardiovasculaire (athérosclérose, thrombose). Le stress est également impliqué dans le déclenchement et/ou la majoration de symptômes digestifs. Le CRF apparaît au centre des mécanismes physiopathologiques des effets du stress sur le tube digestif. Concernant les troubles musculosquelettiques, il est aujourd’hui reconnu que le stress potentialise les effets des sur-sollicitations biomécaniques. Les effets du stress semblent transmis par le système autonome, le système endocrine et le système immunitaire. Enfin, des liens très étroits existent entre les deux axes principaux du stress et le système immunitaire. Ce dernier est informé, par l’intermédiaire des systèmes nerveux autonome et central, de stimuli cognitifs, émotifs et physiques intégrés par le cerveau. En retour, le cerveau reçoit des messages du système immunitaire par l’intermédiaire de neuropeptides hormonaux et de cytokines. Les conséquences pathologiques du stress peuvent résulter d’altérations immunitaires. Le stress, via l’induction d’une transition dans l’équilibre entre lymphocytes TH-1 et TH-2, aurait des effets délétères, dans l’évolution des maladies infectieuses, auto-immunes, inflammatoires et cancéreuses.
Bibliographie
[1] AHOLA K, HONKONEN T, KIVIMÄKI M, VIRTANEN M, ISOMETSÄ E, et coll. Contribution of burnout to the association between job strain and depression: the health 2000 study.
J Occup Environ Med. 2006;
48:1023- 1030
[2] AKERSTEDT T. Psychosocial stress and impaired sleep.
Scand J Work Environ Health. 2006;
32:493501
[3] AMBROGGI F, TURIAULT M, MILET A, DEROCHE-GAMONET V, PARNAUDEAU S, et coll. Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking.
Nat Neurosci. 2009;
12:247- 249
[4] ANACKER C, ZUNSZAIN PA, CARVALHO LA, PARIANTE CM. The glucocorticoid receptor: Pivot of depression and of antidepressant treatment?.
Psychoneuroendocrinology. 2010;
Apr 15. Epub ahead of print;
[5] ANISMAN H, MERALI Z, HAYLEY S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders.
Prog Neurobiol. 2008;
85:1- 74
[6] ANTONI MH, LUTGENDORF SK, COLE SW, DHABHAR FS, SEPHTON SE, et coll. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms.
Nat Rev Cancer. 2006;
6:240- 248
[7] APTEL M. De l’épidémiologie à la physiopathologie des TMS : le modèle de Bruxelles un référentiel intégrateur.
In: FOUQUET B, LASFARGUES G, ROQUELAURE Y, HERISSON C (eds), editors.
Collection Pathologie locomotrice et de médecine orthopédique, Masson édition;
Paris:2007 ;
5162
[8] APTEL M, CNOCKAERT JC. Liens entre les troubles musculo-squelettiques du membre supérieur et le stress.
BTS Newsletter. 2002;
19-20:57- 63
[9] ARMON G, SHIROM A, SHAPIRA I, MELAMED S. On the nature of burnout-insomnia relationships: a prospective study of employed adults.
J Psychosom Res. 2008;
65:5- 12
[10] BAUNE B. Conceptual challenges of a tentative model of stress-induced depression.
PloS One. 2009;
4:e4266
[11] BELKIC KL, LANDSBERGIS PA, SCHNALL PL, BAKER D. Is job strain a major source of cardiovascular disease risk?.
Scand J Work Environ Health. 2004;
30:85- 128
[12] BODOSI B, OBÁL F JR, GARDI J, KOMLÓDI J, FANG J, KRUEGER JM. An ether stressor increases REM sleep in rats: possible role of prolactin.
Am J Physiol Regul Integr Comp Physiol. 2000;
279:R1590- R1598
[13] BONDE JP. Psychosocial factors at work and risk of depression: a systematic review of the epidemiological evidence.
Occup Environ Med. 2008;
65:438445
[14] BRUNNER EJ, HEMINGWAY H, WALKER BR, PAGE M, CLARKE P, et coll. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: nested case-control study.
Circulation. 2002;
106:26592665
[15] CARRICO AW, ANTONI MH. Effects of psychological interventions on neuroendocrine hormone regulation and immune status in HIV-positive persons: a review of randomized controlled trials.
Psychosom Med. 2008;
70:575- 584
[16] CASO JR, LEZA JC, MENCHÉN L. The effects of physical and psychological stress on the gastro-intestinal tract: lessons from animal models.
Curr Mol Med. 2008;
8:299312
[17] CHANDOLA T, BRUNNER E, MARMOT M. Chronic stress at work and the metabolic syndrome: prospective study 2006.
BMJ. 2006;
521- 525. Epub 2006 Jan 20
[18] CHANG FC, OPP MR. Corticotropin-releasing hormone (CRH) as a regulator of waking.
Neurosci Biobehav Rev. 2001;
25:445453
[19] CHAOULOFF F, BERTON O, MORMÈDE P. Serotonin and stress.
Neuropsychopharmacology. 1999;
21(2 Suppl) :28S32S
[20] CHIDA Y, HAMER M, WARDLE J, STEPTOE A. Do stress-related psychosocial factors contribute to cancer incidence and survival?.
Nat Clin Pract Oncol. 2008;
5:466- 475
[21] DALLMAN MF, PECORARO N, AKANA SF, LA FLEUR SE, GOMEZ F, HOUSHYAR H, et coll. Chronic stress and obesity: a new view of “comfort food”.
Proc Natl Acad Sci USA. 2003;
100:11696- 11701
[22] DANTZER R, O’CONNOR JC, FREUND GG, JOHNSON RW, KELLEY KW. From inflammation to sickness and depression: when the immune system subjugates the brain.
Nat Rev Neurosci. 2008;
9:46- 56
[23] DE JONG IE, DE KLOET ER. Glucocorticoids and vulnerability to psychostimulant drugs: toward substrate and mechanism.
Ann N Y Acad Sci. 2004;
1018:192- 198
[24] DE KLOET ER. Hormones and the stressed brain.
Ann N Y Acad Sci. 2004;
1018:1- 15
[25] DHABHAR FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology.
Neuroimmunomodulation. 2009;
16:300- 317
[26] DISCHER D, DONG C, FREDBERG JJ, GUILAK F, INGBER D, et coll. Biomechanics: cell research and applications for the next decade.
Ann Biomed Eng. 2009;
37:847- 859
[27] FOX CM, HARPER AP, HYNER GC, LYLE RM. Loneliness, emotional repression, marital quality, and major life events in women who develop breast cancer.
J Community Health. 1994;
19:467- 482
[28] FOX HC, HONG KI, SIEDLARZ K, SINHA R. Enhanced sensitivity to stress and drug/alcohol craving in abstinent cocaine-dependent individuals compared to social drinkers.
Neuropsychopharmacology. 2008;
33:796- 805
[29] FRICK LR, RAPANELLI M, BUSSMANN UA, KLECHA AJ, ARCOS ML, et coll. Involvement of thyroid hormones in the alterations of T-cell immunity and tumor progression induced by chronic stress.
Biol Psychiatry. 2009;
65:935- 942
[30] FUCHS E, CZÉH B, KOLE MH, MICHAELIS T, LUCASSEN PJ. Alterations of neuroplasticity in depression: the hippocampus and beyond.
Eur Neuropsychopharmacol. 2004;
14(Suppl 5) :S481- S490
[31] GISBERT JP, CALVET X. Review article: Helicobacter pylori-negative duodenal ulcer disease.
Aliment Pharmacol Ther. 2009;
30:791815
[32] GLASER R. Stress-associated immune dysregulation and its importance for human health: a personal history of psychoneuroimmunology.
Brain Behav Immun. 2005;
19:3- 11
[33] GLASER R, MACCALLUM RC, LASKOWSKI BF, MALARKEY WB, SHERIDAN JF, KIECOLT-GLASER JK. Evidence for a shift in the Th-1 to Th-2 cytokine response associated with chronic stress and aging.
J Gerontol A Biol Sci Med Sci. 2001;
56:477- 482
[34] GODBOUT JP, GLASER R. Stress-induced immune dysregulation: implications for wound healing, infectious disease and cancer.
J Neuroimmune Pharmacol. 2006;
1:421427
[35] GODIN I, KITTEL F, COPPIETERS Y, SIEGRIST J. A prospective study of cumulative job stress in relation to mental health.
BMC Public Health. 2005;
15:67
[36] GRAHAM J, RAMIREZ A, LOVE S, RICHARDS M, BURGESS C. Stressful life experiences and risk of relapse of breast cancer: observational cohort study.
BMJ. 2002;
324:1420- 1424
[37] GRIPPO AJ, JOHNSON AK. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models.
Stress. 2009;
12:1- 21
[38] GUÉ M, BONBONNE C, FIORAMONTI J, MORÉ J, DEL RIO-LACHÈZE C, COMÉRA C, BUÉNO L. Stress-induced enhancement of colitis in rats: CRF and arginine vasopressin are not involved.
Am J Physiol. 1997;
272:G84- G91
[39] GUO G, JIA KR, SHI Y, LIU XF, LIU KY, et coll. Psychological stress enhances the colonization of the stomach by Helicobacter pylori in the BALB/c mouse.
Stress. 2009;
12:478- 485
[40] HEAD J, STANSFELD SA, SIEGRIST J. The psychosocial work environment and alcohol dependence: a prospective study.
Occup Environ Med. 2004;
61:219- 224
[41] JOHANSSON H, WINDHORST U, DJUPSJÖBACKA M, PASSATORE M. Chronic work-related myalgia: Neuromuscular mechanisms behind work-related chronic muscle pain syndromes.
In: JOHANSSON H, WINDHORST U, DJUPSJÖBACKA M, PASSATORE M (eds), editors.
Gvale University Press;
Gvale: Sweden.
2003;
310 p
[42] JOUVET M. Sleep and serotonin: an unfinished story.
Neuropsychopharmacology. 1999;
21:24S- 27S
[43] KAPRARA A, PAZAITOU-PANAYIOTOU K, KORTSARIS A, CHATZAKI E. The corticotropin releasing factor system in cancer: expression and pathophysiological implications.
Cell Mol Life Sci. 2010;
67:1293- 306. Epub 2010 Feb 9
[44] KEITA AV, SÖDERHOLM JD. The intestinal barrier and its regulation by neuroimmune factors.
Neurogastroenterol Motil. 2010;
Apr 9. [Epub ahead of print];
[45] KELLER J, FLORES B, GOMEZ RG, SOLVASON HB, KENNA H, et coll. Cortisol circadian rhythm alterations in psychotic major depression.
Biol Psychiatry. 2006;
60:275- 281
[46] KEMENY ME, SCHEDLOWSKI M. Understanding the interaction between psychosocial stress and immune-related diseases: a stepwise progression.
Brain Behav Immun. 2007;
21:1009- 1018
[47] KIECOLT-GLASER JK, GLASER R. Psychoneuroimmunology and cancer: fact or fiction?.
Eur J Cancer. 1999;
35:1603- 1607
[48] KNUTSON KL, SPIEGEL K, PENEV P, VAN CAUTER E. The metabolic consequences of sleep deprivation.
Sleep Med Rev. 2007;
11:163- 178
[49] KUO LE, CZARNECKA M, KITLINSKA JB, TILAN JU, KVETNANSKÝ R, ZUKOWSKA Z. Chronic stress, combined with a high-fat/high-sugar diet, shifts sympathetic signaling toward neuropeptide Y and leads to obesity and the metabolic syndrome.
Ann N Y Acad Sci. 2008;
1148:232- 237
[50] KURATA JH, NOGAWA AN. Meta-analysis of risk factors for peptic ulcer. Nonsteroidal antiinflammatory drugs, Helicobacter pylori, and smoking.
J Clin Gastroenterol. 1997;
24:2- 17
[51] KYROU I, TSIGOS C. Stress mechanisms and metabolic complications.
Horm Metab Res. 2007;
39:430- 438
[52] KYROU I, TSIGOS C. Chronic stress, visceral obesity and gonadal dysfunction.
Hormones. 2008;
7:287- 293
[53] KYROU I, TSIGOS C. Stress hormones: physiological stress and regulation of metabolism.
Curr Opin Pharmacol. 2009;
9:787- 793
[54] LANDFIELD PW, BLALOCK EM, CHEN KC, PORTER NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease.
Curr Alzheimer Res. 2007;
4:205- 212
[55] LILLBERG K, VERKASALO PK, KAPRIO J, TEPPO L, HELENIUS H, KOSKENVUO M. Stressful life events and risk of breast cancer in 10,808 women: a cohort study.
Am J Epidemiol. 2003;
157:415- 423
[56] LOPEZ M, CAUCHI C, SERGI D, AMODIO A, PAOLETTI G, et coll. Psyche and cancer.
Clin Ter. 2010;
161:69- 75
[57] MACFARLANE GJ, PALLEWATTE N, PAUDYAL P, BLYTH FM, COGGON D, et coll. Evaluation of work-related psychosocial factors and regional musculoskeletal pain: results from a EULAR Task Force.
Ann Rheum Dis. 2009;
68:885- 891
[58] MARINELLI M, PIAZZA PV. Interaction between glucocorticoid hormones, stress and psychostimulant drugs.
Eur J Neurosci. 2002;
16:387- 394
[59] MAUNDER RG, LEVENSTEIN S. The role of stress in the development and clinical course of inflammatory bowel disease: epidemiological evidence.
Curr Mol Med. 2008;
8:247- 252
[60] MELCHIOR M, CASPI A, MILNE BJ, DANESE A, POULTON R, MOFFITT TE. Work stress precipitates depression and anxiety in young, working women and men.
Psychol Med. 2007;
37:1119- 1129
[61] MENDLEWICZ J, KERKHOFS M. Sleep electroencephalography in depressive illness. A collaborative study by the World Health Organization.
Br J Psychiatry. 1991;
159:505- 509
[62] MILLER G, CHEN E, COLE SW. Health Psychology: Developing biologically plausible models linking the social world and physical health.
Annual Review of Psychology. 2009;
60:501- 524
[63] MOHR DC. Stress and multiple sclerosis.
J Neurol. 2007;
254(Suppl 2) :II65- II68
[64] MOHR DC, HART SL, JULIAN L, COX D, PELLETIER D. Association between stressful life events and exacerbation in multiple sclerosis: a meta-analysis.
BMJ. 2004;
328:731
[65] NETTERSTRØM B, CONRAD N, BECH P, FINK P, OLSEN O, et coll. The relation between work-related psychosocial factors and the development of depression.
Epidemiol Rev. 2008;
30:118- 132
[66] NIELSEN LS, DANIELSEN KV, SØRENSEN TI. Short sleep duration as a possible cause of obesity: critical analysis of the epidemiological evidence.
Obes Rev. 2010;
mars 24. [Epub ahead of print];
[67] NILSEN KB, SAND T, STOVNER LJ, LEISTAD RB, WESTGAARD RH. Autonomic and muscular responses and recovery to one-hour laboratory mental stress in healthy subjects.
BMC Musculoskelet Disord. 2007;
14:81- 93
[68] OHLMANN KK, O’SULLIVAN MI. The costs of short sleep.
AAOHN J. 2009;
57:381- 385
[69] PARIANTE CM, LIGHTMAN SL. The HPA axis in major depression: classical theories and new developments.
Trends Neurosci. 2008;
31:464- 468
[70] PAWLYK AC, SANFORD LD, BRENNAN FX, MORRISON AR, ROSS RJ. Corticotropin-releasing factor microinjection into the central nucleus of the amygdala alters REM sleep.
Pharmacol Rep. 2006;
58:125- 130
[71] PIAZZA PV, LE MOAL M. Pathophysiological basis of vulnerability to drug abuse: role of an interaction between stress, glucocorticoids, and dopaminergic neurons.
Annu Rev Pharmacol Toxicol. 1996;
36:359- 378
[72] PIAZZA PV, LE MOAL M. The role of stress in drug self-administration.
Trends Pharmacol Sci. 1998;
19:67- 74
[73] PITTENGER C, DUMAN RS. Stress, depression, and neuroplasticity: a convergence of mechanisms.
Neuropsychopharmacology. 2008;
33:88- 109
[74] PUDUVALLI VK, SELLA A, AUSTIN SG, FORMAN AD. Carpal tunnel syndrome associated with interleukin-2 therapy.
Cancer. 1996;
77:1189- 1192
[75] RAISON CL, CAPURON L, MILLER AH. Cytokines sing the blues: inflammation and the pathogenesis of depression.
Trends Immunol. 2006;
27:24- 31
[76] REICHE EM, NUNES SO, MORIMOTO HK. Stress, depression, the immune system, and cancer.
Lancet Oncol. 2004;
5:617- 625
[77] RICHARDSON HN, ZHAO Y, FEKETE EM, FUNK CK, WIRSCHING P, et coll. MPZP: a novel small molecule corticotropin-releasing factor type 1 receptor (CRF1) antagonist.
Pharmacol Biochem Behav. 2008;
88:497- 510
[78] ROSAS-BALLINA M, TRACEY KJ. The neurology of the immune system: neural reflexes regulate immunity.
Neuron. 2009;
64:28- 32
[79] ROSMOND R. Role of stress in the pathogenesis of the metabolic syndrome.
Psychoneuroendocrinology. 2005;
30:1- 10
[80] ROTHMAN SM, MATTSON MP. Adverse stress, hippocampal networks, and Alzheimer’s disease.
Neuromolecular Med. 2010;
12:56- 70
[81] SAPOLSKY RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders.
Arch Gen Psychiatry. 2000;
57:925- 935
[82] SAPOLSKY RM, KREY LC, MCEWEN BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis.
Endocr Rev. 1986;
7:284- 301
[83] SCHRAUB S, SANCHO-GARNIER H, VELTEN M. Should psychological events be considered cancer risk factors?.
Rev Epidemiol Sante Publique. 2009;
57:113- 123
[84] SEPHTON S, SPIEGEL D. Circadian disruption in cancer: a neuroendocrine-immune pathway from stress to disease?.
Brain Behav Immun. 2003;
17:321- 328
[85] SEPHTON SE, SAPOLSKY RM, KRAEMER HC, SPIEGEL D. Diurnal cortisol rhythm as a predictor of breast cancer survival.
J Natl Cancer Inst. 2000;
92:994- 1000
[86] SIEGRIST J. Chronic psychosocial stress at work and risk of depression: evidence from prospective studies.
Eur Arch Psychiatry Clin Neurosci. 2008;
258(Suppl 5) :115- 119
[87] SIEGRIST J, RÖDEL A. Work stress and health risk behavior.
Scand J Work Environ Health. 2006;
32:473- 481
[88] SOTIROPOULOS I, CERQUEIRA JJ, CATANIA C, TAKASHIMA A, SOUSA N, ALMEIDA OF. Stress and glucocorticoid footprints in the brain-the path from depression to Alzheimer’s disease.
Neurosci Biobehav Rev. 2008;
32:1161- 1173
[89] SOUÊTRE E, SALVATI E, BELUGOU JL, PRINGUEY D, CANDITO M, et coll. Circadian rhythms in depression and recovery: evidence for blunted amplitude as the main chronobiological abnormality.
Psychiatry Res. 1989;
28:263- 278
[90] SPECIO SE, WEE S, O’DELL LE, BOUTREL B, ZORRILLA EP, KOOB GF. CRF(1) receptor antagonists attenuate escaladed cocaine self-administration in rats.
Psychopharmacology. 2008;
196:473- 482
[91] SPIEGEL K, LEPROULT R, VAN CAUTER E. Impact of sleep debt on metabolic and endocrine function.
Lancet. 1999;
354:1435- 1439
[92] SPIEGEL K, TASALI E, LEPROULT R, VAN CAUTER E. Effects of poor and short sleep on glucose metabolism and obesity risk.
Nat Rev Endocrinol. 2009;
5:253- 261
[93] STÖHR T, SZURAN T, WELZL H, PLISKA V, FELDON J, PRYCE CR. Lewis/Fischer rat strain differences in endocrine and behavioural responses to environmental challenge.
Pharmacol Biochem Behav. 2000;
67:809- 819
[94] TACHÉ Y, BONAZ B. Corticotropin-releasing factor receptors and stress-related alterations of gut motor function.
J Clin Invest. 2007;
117:33- 40
[95] TACHÉ Y, BRUNNHUBER S. From Hans Selye’s discovery of biological stress to the identification of corticotropin-releasing factor signaling pathways: implication in stress-related functional bowel diseases.
Ann N Y Acad Sci. 2008;
1148:29- 41
[96] TEEGARDEN SL, BALE TL. Effects of stress on dietary preference and intake are dependent on access and stress sensitivity.
Physiol Behav. 2008;
93:713723
[97] TWELLAAR M, WINANTS Y, HOUKES I. How healthy are Dutch general practitioners? Self-reported (mental) health among Dutch general practitioners.
Eur J Gen Pract. 2008;
14:4- 9
[98] URSIN R. Serotonin and sleep.
Sleep Med Rev. 2002;
6:55- 69
[99] VAN REETH O, WEIBEL L, SPIEGEL K, LEPROUT R, DUGOVIC C, MACCARI S. Interactions between stress and sleep: from basic research to clinical situations.
Sleep Med Rev. 2000;
4:201- 219
[100] VÁZQUEZ-PALACIOS G, RETANA-MÁRQUEZ S, BONILLA-JAIME H, VELÁZQUEZ-MOCTEZUMA J. Stress-induced REM sleep increase is antagonized by naltrexone in rats.
Psychopharmacology (Berl). 2004;
171:186- 190
[101] VELIN D, MICHETTI P. Immunology of Helicobacter pylori infection.
Digestion. 2006;
73 :116- 123
[102] VERE CC, STREBA CT, STREBA LM, IONESCU AG, SIMA F. Psychosocial stress and liver disease status.
World J Gastroenterol. 2009;
15:2980- 2986
[103] WANG J, LI S. Corticotropin-releasing factor family and its receptors: tumor therapeutic targets?.
Biochem Biophys Res Commun. 2007;
362:785- 788
[104] WEBSTER MARKETON JI, GLASER R. Stress hormones and immune function.
Cell Immunol. 2008;
252:16- 26
[105] WIRZ-JUSTICE A. Biological rhythm disturbances in mood disorders.
Int Clin Psychopharmacol. 2006;
21(Suppl 1) :S11- S15
[106] YEE JR. Health and sociality: exploring bidirectionnal relationships between behavior and physiology from a systems perspective. PhD dissertation.
The University of Chicago;
Chicago: Illinois.
2008;
Available from: ProQuest Digital dissertations. Accessed November 24, 2009, Publication number: AAT 3338529;
→ Aller vers SYNTHESE