Pharmacodépendances et mésusages
2012
12-
Pharmacodépendance : mécanismes neurobiologiques
Les propriétés renforçantes d’une substance psychoactive sont à l’origine du maintien de sa consommation. Elles peuvent être de nature positive ou négative. Un renforcement positif correspond à un effet direct de la substance sur les voies cérébrales hédoniques (circuit de la récompense). Le renforcement négatif, par le maintien de l’usage, permet par exemple d’éviter les effets désagréables du sevrage. Dans les deux cas, la poursuite de la consommation peut s’accompagner d’une augmentation de la fréquence d’administration.
Ces deux versants peuvent se retrouver dans le mésusage et la pharmacodépendance aux médicaments psychotropes. Ce mésusage correspond à une consommation non-conforme et/ou problématique, qu’il soit à visée thérapeutique, récréative, toxicomaniaque ou pour gérer une dépendance.
Les médicaments psychotropes peuvent être utilisés seuls ou en association avec d’autres substances psychoactives, licites ou illicites. Cette association peut être destinée à une recherche d’effets plus importants (synergie), ou à atténuer des effets de descente liés à la prise de drogues (qui se caractérisent par différents symptômes : dysphorie, fatigue, irritabilité, anxiété…).
Les connaissances sur les mécanismes qui sous-tendent les processus d’addiction ont beaucoup progressé au cours des dernières années. Les données acquises permettent aujourd’hui d’appréhender de façon plus concrète les risques de mésusage et de pharmacodépendance aux médicaments psychotropes, d’autant plus si les propriétés pharmacocinétiques et pharmacodynamiques des différents médicaments psychotropes sont bien définies.
Neurotransmetteurs et circuits impliqués dans la neurobiologie des addictions
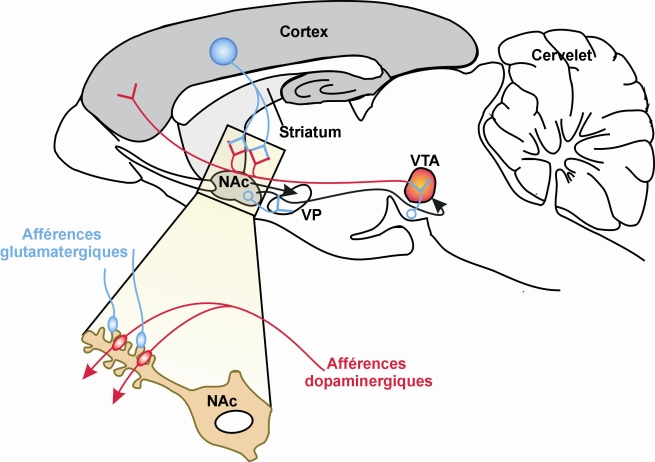
Toutes les substances addictives (y compris le tabac et l’alcool) agissent directement ou indirectement sur les mêmes réseaux de neurones du système nerveux central, le système mésocorticolimbique, et conduisent à une augmentation extracellulaire de dopamine. Ce système est formé de neurones dopaminergiques (neurones qui synthétisent la dopamine comme neurotransmetteur). Les corps cellulaires de ces neurones sont situés dans l’aire tegmentale ventrale (ATV) et leurs axones atteignent le noyau accumbens, le tubercule olfactif, le cortex frontal et l’amygdale.
Schématiquement, la voie dopaminergique peut être activée de deux façons :
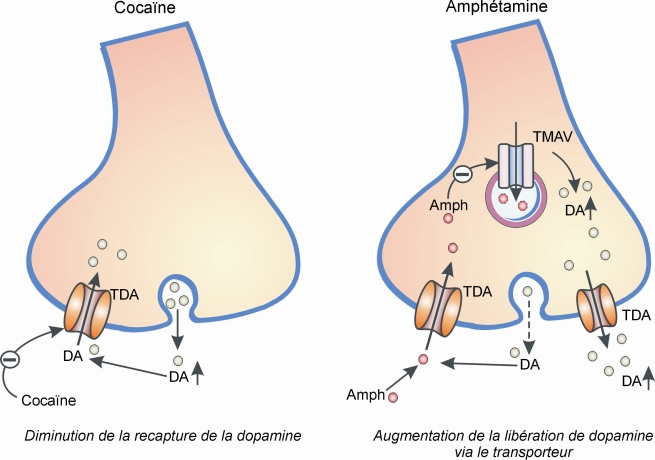
• une activation directe (figure 12.1

) par des substances qui, soit favorisent la libération de dopamine (amphétamine, methamphétamine, phentermine…) soit inhibent sa recapture (cocaïne, méthylphénidate, bupropion…) au niveau des terminaisons dans le noyau accumbens ;
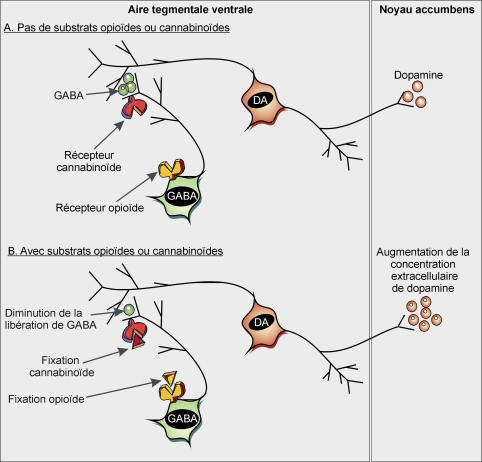
• une activation indirecte (figure 12.2
) par des substances (opioïdes tels que morphine, rémifentanyl, buprénorphine, cannabis) qui lèvent l’inhibition du fonctionnement de la voie mésocorticolimbique au niveau des neurones dopaminergiques de l’ATV. Cette inhibition est normalement assurée par des interneurones GABAergiques présents dans l’ATV. L’activation des récepteurs opioïdes ou cannabinoïdes présents sur ces interneurones GABAergiques permet de diminuer la libération du neurotransmetteur GABA, et donc de diminuer l’inhibition du neurone dopaminergique, ce qui conduit à une augmentation de la libération de dopamine dans le noyau accumbens.
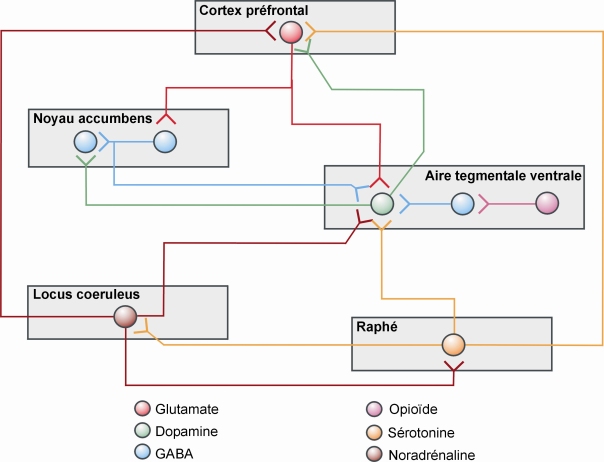
La dopamine est certainement une molécule centrale dans la mise en place des processus de dépendance, mais d’autres neurotransmetteurs et neuromodulateurs sont également impliqués, les principaux étant le GABA, le glutamate, la sérotonine, la noradrénaline et les peptides opioïdes. Les informations reçues sont traitées par un ensemble complexe de circuits neuronaux (figure 12.3
). L’implication de ces neurotransmetteurs peut être indirecte, via les interactions qu’ils peuvent avoir avec le système dopaminergique, et/ou directe, avec des modifications spécifiques de ces systèmes dans les processus de dépendance.
Évaluation des effets renforçants d’une substance psychoactive : modèles prédictifs précliniques
Le mésusage est souvent associé à un mode de consommation différent de celui prescrit (modification de la voie d’administration, augmentation des quantités absorbées…) et qui aboutit à augmenter les concentrations cérébrales du produit psychotrope (Sussman et coll., 2006
). Il est bien établi que la concentration plasmatique d’une substance addictive et la vitesse à laquelle elle atteint la circulation sanguine conditionnent les effets pharmacologiques, et en particulier les effets renforçants positifs. Ces effets peuvent être explorés expérimentalement chez l’animal.
En France, l’évaluation de l’installation potentielle d’une pharmacodépendance chez l’animal de laboratoire fait partie des recommandations préconisées pour le développement de toute molécule active sur le système nerveux central
1
L’Agence européenne des médicaments (European Medicines Agency, EMA) a émis des recommandations aux demandeurs d’AMM de substances capables d’agir dans le système nerveux central, afin qu’ils réalisent des études précliniques permettant d’évaluer le potentiel addictif des nouvelles molécules, et que les résultats soient présentés dans les dossiers (EMEA/CHMP/SWP/94227/2004). Ceci concerne par voie de conséquence les demandes émanant de la France.
. Elle a pour but, en particulier, d’estimer le potentiel d’abus. Les études précliniques permettent d’évaluer ce potentiel très tôt (dès l’exposition
in utero), d’utiliser une large gamme de doses, et de suivre des animaux traités sur de longues périodes de vie. Plusieurs modèles comportementaux sont actuellement utilisés (revues dans Balster et Bigelow, 2003
; Ator et Griffiths, 2003
).
L’addiction résulte de mécanismes complexes et multifactoriels, faisant intervenir la rencontre d’un individu avec un produit, dans un contexte bien défini. De par sa complexité, il n’existe pas de modèle animal parfait répliquant point par point la pathologie humaine. Néanmoins différents modèles existent, fondés sur les effets positifs des substances (auto-administration, préférence de place conditionnée, modèle de discrimination), ou sur leurs effets négatifs (sevrage, aversion de place conditionnée). Il existe également des modèles fondés sur l’escalade des doses, ou encore des modèles de rechute (auto-administration, préférence de place conditionnée).
Modèle de discrimination
Le modèle de discrimination repose sur trois phases distinctes. La première consiste en l’acquisition d’un comportement : l’animal, le plus souvent un rat, est placé dans une cage en présence de deux leviers (A et B) qu’il peut actionner pour obtenir une récompense (nourriture ou eau sucrée, par exemple). Lors de la deuxième phase (entraînement), le rat apprend à associer les effets que lui procure la substance à l’étude à un comportement spécifique : la substance lui est injectée et seul un des deux leviers est actif (levier A par exemple) puis, de façon alternée, on lui administre le solvant (placebo) et seul l’autre levier (B) est actif. Au cours de cette phase, le travail demandé à l’animal augmente progressivement en passant d’un FR1 (fixed ratio, 1 appui = 1 récompense) à un FR3 (3 appuis = 1 récompense), puis FR5, et FR10. Une fois le comportement de l’animal stabilisé, on pourra alors passer à la troisième phase du modèle : le test. Au cours de cette phase, les deux leviers sont inactivés, et on injecte à l’animal la molécule testée (soit une autre molécule que celle utilisée lors de l’entraînement, soit la même molécule mais à une dose différente). Si la préférence de l’animal va vers le levier A, cela indiquera que la substance testée (ou la nouvelle dose utilisée) procure à l’animal des effets subjectifs analogues à ceux de la substance (ou de la dose) de référence. En revanche, si sa préférence va vers le levier B, on pourra conclure que les effets sont semblables à ceux produits par le placebo, et si l’animal ne montre aucune préférence pour l’un ou l’autre levier, on conclura que les effets sont autres.
Modèle d’auto-administration
Le modèle d’auto-administration prend en compte une variable mesurable qui s’apparente à la prise compulsive de substance psychotrope observable chez des patients dépendants. Ce modèle permet de mesurer la valeur hédonique de la substance qui se manifeste par un comportement actif de l’animal en vue de se la procurer. Les substances sont généralement introduites par voie intraveineuse à l’aide d’un cathéter implanté à demeure dans la veine jugulaire. L’animal est placé dans une cage d’expérimentation et relié à un système d’injection qu’il déclenchera à volonté par appui sur un levier. Au cours des premières séances, l’animal activera fortuitement le levier et recevra une injection. Si les effets sont agréables, le rat va répéter cette action afin de s’injecter de plus en plus de produit.
Préférence de place conditionnée
Parmi les diverses approches d’évaluation des effets renforçants d’une molécule psychoactive, la procédure de conditionnement spatial ou de préférence de place conditionnée est l’une des plus intéressantes. Il ne s’agit pas à proprement parler d’un modèle d’addiction car il n’est pas donné à l’animal la possibilité de s’auto-administrer une substance, mais il permet d’évaluer l’intensité du souvenir, de la valeur hédonique que les effets d’une substance procurent à l’animal. Dans ces expériences, l’animal est placé dans une cage présentant plusieurs compartiments qui se distinguent par la couleur des parois, la texture du sol et par différentes odeurs. Au cours d’une première séance, l’animal va explorer librement les divers compartiments. Dans une deuxième étape, l’animal apprend à associer les effets de la substance à un environnement spécifique : il sera confiné dans un compartiment particulier après l’administration de celle-ci, puis le lendemain dans un autre compartiment après l’administration du solvant. Cette étape est répétée plusieurs jours. Au cours de la troisième phase, l’animal (ne recevant plus d’injection) est réintroduit dans la cage avec libre accès aux différents compartiments. Sa préférence pour le compartiment associé à la substance testée révélera la valeur renforçante du produit. À l’inverse, les effets aversifs d’une substance seront révélés par l’évitement du compartiment associé à la molécule.
Dépendance physique et sevrage
La dépendance physique se traduit par la survenue de symptômes spécifiques comportementaux et somatiques, qui caractérisent le syndrome de sevrage. Elle résulte des mécanismes d’adaptation de l’organisme à une consommation prolongée. Elle peut être accompagnée d’une accoutumance (ou tolérance), c’est-à-dire une nécessité d’augmenter les doses pour éprouver un même effet.
Sensibilisation comportementale
Le terme de sensibilisation réfère à l’augmentation d’une réponse comportementale suite à l’administration répétée d’un composé, par comparaison à celle observée après administration aiguë. Dans le cas des psychostimulants, une sensibilisation du comportement locomoteur chez le rongeur peut être facilement mise en évidence en plaçant l’animal dans une cage munie de cellules photoélectriques. Le nombre de coupures des faisceaux permet de quantifier l’activité locomotrice et ainsi les effets du produit étudié. Cette sensibilisation serait associée à des modifications morphologiques des neurones (avec en particulier des augmentations de la densité des épines dendritiques) et à l’altération à long terme de l’activité de différentes voies de neurotransmission (dopamine, sérotonine, glutamate, par exemple).
Pour résumer, même si ces modèles présentent des limites, ils ont des avantages uniques par rapport aux études cliniques (effet-dose, études longitudinales à long terme…), et sont prédictifs de ce qui peut être observé chez l’Homme. Cependant, lors de l’extrapolation des résultats de l’animal à l’Homme, certaines différences inter-espèces doivent être prises en considération, en particulier le passage de la barrière hémato-encéphalique et le métabolisme des molécules mères. En effet, le passage d’un psychotrope du sang vers le cerveau dépend de la capacité de la substance à traverser une barrière physique composée de cellules endothéliales jointives exprimant sur leur membrane des transporteurs d’efflux et d’influx qui sont capables de moduler les concentrations cérébrales de psychotrope. Cette barrière peut également être une barrière métabolique due à l’expression dans ces cellules endothéliales d’enzymes du métabolisme des médicaments, métabolisme qui peut également se faire au niveau hépatique. La caractérisation des métabolites est importante, les propriétés renforçantes pouvant venir d’un métabolite et non de la molécule mère. Tout métabolite actif chez l’Homme doit être étudié chez l’animal, afin de disposer des meilleures prédictibilités possibles.
Liens entre mésusages et pharmacodépendances
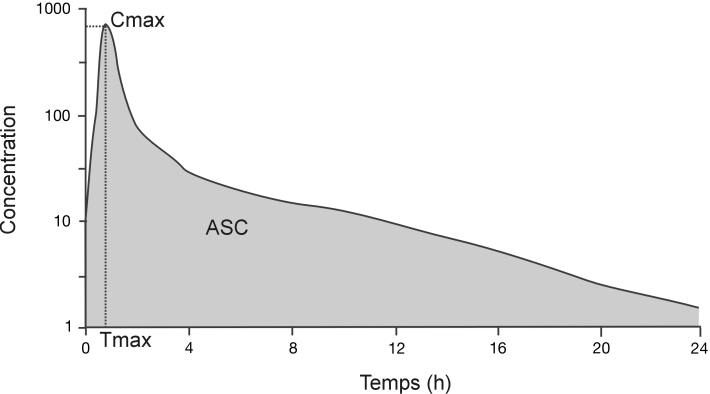
Selon les principes fondamentaux de la pharmacologie, la quantité et la vitesse auxquelles un composé atteint la circulation sanguine, permettant sa distribution dans l’organisme et donc l’accès à ses sites d’action, conditionnent ses effets pharmacologiques. D’un point de vue pharmacocinétique, les effets pharmacologiques d’une molécule dépendent soit de sa concentration maximale (Cmax, effet pic), soit de son exposition systémique reflétée par ses concentrations sanguines en fonction du temps (aire sous la courbe) (figure 12.4
).
Importance de la voie d’administration
Le profil pharmacocinétique d’une substance dépend de la voie d’administration. Le facteur de biodisponibilité définissant la fraction d’une dose qui atteint la circulation sanguine est par définition égal à 100 % pour une administration par voie intra-vasculaire (intraveineuse, intra-artérielle), la substance étant injectée directement dans la circulation, et le Tmax (temps nécessaire pour atteindre la Cmax) est observé très rapidement (de l’ordre de 3 à 5 min) après administration. En revanche, une substance administrée par voie extra-vasculaire (orale, intramusculaire, sous-cutanée…) présente une phase d’absorption avec une biodisponibilité pouvant varier de 0 à 100 % et un Tmax en général plus long (souvent supérieur à 30 min).
La voie d’administration conditionne la dépendance aux substances psychotropes, comme cela a été montré pour la cocaïne qui présente des effets addictifs plus importants lorsqu’elle est fumée plutôt que sniffée. La Cmax de la cocaïne fumée est au moins deux fois supérieure à celle obtenue après consommation par voie nasale (sniff) (Cone, 1995
). Les effets addictifs de la cocaïne (et aussi d’autres produits) sont donc très probablement liés à l’effet pic plutôt qu’à son exposition systémique. Cet effet pic se caractérise par le « flash » rapporté par les consommateurs et peut être corrélé à une libération de dopamine (neurotransmetteur du plaisir) rapide et importante.
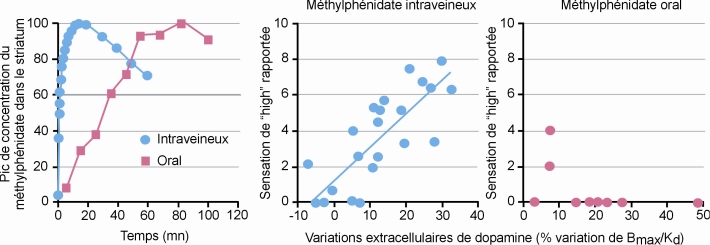
Des études en neuroimagerie chez l’Homme montrent que la libération rapide de dopamine obtenue après injection intraveineuse de méthylphénidate (composé capable d’augmenter les concentrations de dopamine par blocage des systèmes de recapture) est associée à des effets renforçants de ce composé. En revanche, une augmentation de dopamine de même amplitude mais plus lente, obtenue après administration orale d’une dose permettant d’obtenir la même concentration cérébrale de méthylphénidate qu’après administration intraveineuse, n’est pas perçue comme renforçante (Volkow et coll., 1999
; Volkow et Swanson, 2003
) (figure 12.5
). D’un point de vue mécanistique, le méthylphénidate par voie intraveineuse est capable de mimer les décharges phasiques rapides des neurones dopaminergiques (décharges essentielles pour les effets renforçants), alors que par voie orale il mime les décharges toniques de ces neurones (effets expliquant l’effet thérapeutique) (Swanson et Volkow, 2003
). L’importance de la voie d’administration dans le potentiel de propriétés renforçantes du méthylphénidate a également été mise en évidence dans des modèles précliniques : auto-administration, préférence de place conditionnée, sensibilisation comportementale, discrimination (Sellings et coll., 2006
; Botly et coll., 2008
; McGovern et coll., 2011
).
Bien que rare (Berman et coll., 2009
), une pharmacodépendance au méthylphénidate apparaîtra essentiellement après un mode d’utilisation qui permet une libération rapide de dopamine dans le cerveau (haut dosage, en intranasal ou injection intraveineuse essentiellement) comme peut le faire la cocaïne (Sussman et coll., 2006
). Des différences pharmacocinétiques importantes existent néanmoins entre ces deux produits. La clairance du méthylphénidate dans le cerveau est plus lente (90 min), avec un pic de concentration pouvant se maintenir entre 15-20 min, alors que la clairance de la cocaïne est de 20 min, avec un pic de concentration se maintenant seulement entre 2-4 min (Svetlov et coll., 2007
).
Importance de la vitesse d’injection
Plusieurs résultats obtenus aussi bien chez l’Homme que chez l’animal montrent également l’importance de la vitesse d’injection par voie intraveineuse, cette vitesse conditionnant la rapidité d’accès aux sites d’action des composés. Ainsi, injecter chez le rat des doses similaires de cocaïne par voie intraveineuse en 1, 5, 25 ou 100 secondes entraîne des réponses différentes tant au niveau de la cinétique de libération de dopamine, que de l’activation de gènes précoces impliqués dans les phénomènes d’addiction et qui sont corrélés à des variations de réponses comportementales (auto-administration, sensibilisation comportementale) (Samaha et coll., 2004
; Samaha et Robinson, 2005
; Schindler et coll., 2009
). Les effets sont plus prononcés lorsque l’injection est rapide. Ces résultats ont été confirmés chez le singe (Woolverton et Wang, 2004
). Chez l’Homme, l’injection intraveineuse d’une même dose d’oxycodone, un analgésique opiacé, en 2, 15, 30, 60 ou 90 min entraîne également des effets très différents, puisque seuls les volontaires recevant le produit en 2 ou 15 min décrivent des effets renforçants (Comer et coll., 2009
).
Pharmacodépendance aux médicaments psychotropes partageant les cibles moléculaires de la cocaïne
Le mécanisme d’action de la cocaïne est bien défini ; elle bloque les systèmes de recapture de la dopamine, de la noradrénaline et de la sérotonine, permettant ainsi d’augmenter les concentrations synaptiques de ces neurotransmetteurs. Ces systèmes de recapture sont également les cibles moléculaires des médicaments indiqués dans le trouble déficit de l’attention avec hyperactivité et des antidépresseurs.
Médicaments pour le trouble déficit de l’attention avec hyperactivité
Actuellement, les médicaments indiqués dans le traitement du trouble déficit de l’attention avec hyperactivité (TDAH) sont des psychostimulants, en particulier le méthylphénidate (Ritaline®) ou encore l’atomoxétine (Strattera®).
Méthylphénidate
Comparé à la cocaïne qui est capable de bloquer trois systèmes de recapture de neurotransmetteurs, le méthylphénidate n’inhibe que deux d’entre eux : dopamine et noradrénaline.
Au niveau moléculaire, les études transcriptomiques menées chez les rongeurs ont montré des régulations de l’expression génique après administration de cocaïne ou de méthylphénidate. L’expression de facteurs de transcription (zif 268, c-fos), de facteurs de plasticité synaptique (Homer 1a) et de neuropeptides (enképhaline, substance P, dynorphine) est modifiée (augmentation dans la majorité des cas) dans le striatum, une structure jouant un rôle clé dans la neurobiologie des addictions (Yano et Steiner, 2005
et 2007
). Néanmoins, la majorité des effets induits par le méthylphénidate sont observés après administration par des voies permettant des concentrations cérébrales élevées (i.p., s.c., i.v.)
2
i.p. : intrapéritonéale ; s.c. : sous-cutanée ; i.v. : intraveineuse
, et les comparaisons entre cocaïne et méthylphénidate montrent que ce dernier induit moins de neuroadaptations (modifications d’expression de gènes). Cette différence pourrait provenir de l’incapacité du méthylphénidate à stimuler la neurotransmission sérotoninergique (Yano et Steiner, 2007
).
Au niveau anatomique, les neurones striataux reçoivent d’une part des afférences glutamatergiques issues du cortex cérébral, informatrices du contexte, et, d’autre part, des afférences dopaminergiques originaires de l’aire tegmentale ventrale (ATV) dont l’activation peut être considérée comme un signal de récompense. Le striatum est donc une structure cérébrale qui permet l’association entre un contexte et un signal renforçant, et cet apprentissage associatif aboutit à des adaptations comportementales. Dans les neurones striataux, les épines dendritiques qui intègrent les signaux glutamatergiques et dopaminergiques (figure 12.6
), sont des éléments cellulaires-clés dans l’étude des mécanismes qui sous-tendent la consolidation de la trace mnésique.
Chez les rongeurs, l’administration chronique de méthylphénidate augmente la densité des épines dendritiques des neurones exprimant les récepteurs dopaminergiques D1 dans le
core et le
shell du striatum ventral (noyau accumbens), ainsi que des neurones exprimant les récepteurs D2 dans le
shell. En revanche, la cocaïne augmente les épines dendritiques des neurones exprimant les récepteurs D1 et D2 à la fois dans le
shell et le
core du noyau accumbens (Kim et coll., 2009
). Les neuroadaptations induites par la cocaïne apparaissent donc plus importantes que celles induites par le méthylphénidate.
Enfin, les études comportementales montrent que différentes espèces animales développent un comportement d’auto-administration (rat, chien, singe) et une préférence de place conditionnée (rat) après administration intraveineuse de méthylphénidate (Sellings et coll., 2006
; Botly et coll., 2008
). Cependant, des contradictions existent concernant l’impact d’une pré-exposition à ce médicament. Certaines études montrent une attirance des animaux plus importante vers d’autres drogues (en particulier la nicotine et la cocaïne) (Schenk et Izenwasser, 2002
; Wooters et coll., 2008
), alors que d’autres, au contraire, mettent en évidence un effet protecteur. Cette divergence dans les résultats semble essentiellement liée à l’âge des animaux. En effet, une exposition au méthylphénidate à l’âge adulte augmente l’auto-administration de cocaïne (Schenk et Izenwasser, 2002
) et la préférence de place conditionnée au méthylphénidate (Meririnne et coll., 2001
), mais reste sans effet sur la sensibilisation comportementale (Suzuki et coll., 2007
). En revanche, une exposition à l’adolescence semble capable de diminuer l’appétence pour la cocaïne (Thanos et coll., 2007
; Ferguson et Boctor, 2010
) et ne modifie pas la consommation d’éthanol à l’âge adulte (Soeters et coll., 2008
).
Association méthylphénidate et inhibiteur de recapture de la sérotonine
La principale différence en termes de cibles pharmacologiques entre le méthylphénidate et la cocaïne étant l’inhibition du système de recapture de la sérotonine, l’association du méthylphénidate avec un inhibiteur de ce système (comme la fluoxétine) pourrait permettre de mimer les effets de la cocaïne. Il existe effectivement une potentialisation des effets du méthylphénidate par la fluoxétine (antidépresseur inhibiteur spécifique de la recapture de sérotonine ou ISRS, commercialisé sous la spécialité Prozac®), aussi bien au niveau du comportement (stéréotypies, locomotion), que des modifications d’expression de gènes (
c-fos,
zif 268) (Steiner et coll., 2010
; Van Waes et coll., 2010
).
Atomoxétine
L’atomoxétine (Strattera®)
3
L’atomoxetine n’est actuellement pas commercialisée en France.
est un inhibiteur sélectif du système de recapture de la noradrénaline, sans affinité pour les systèmes de recapture de la dopamine et de la sérotonine (Bymaster et coll., 2002
) et utilisé pour le traitement de l’hyperactivité avec troubles attentionnels de l’enfant. L’injection d’atomoxétine chez le rat n’entraîne pas de libération de dopamine dans le striatum (Bymaster et coll., 2002
), ceci pouvant expliquer l’absence d’effets renforçants de cette molécule (Gasior et coll., 2005
). Par ailleurs, chez des rats habitués à s’auto-administrer de la cocaïne, si on remplace ce produit par le méthylphénidate, les animaux maintiennent un comportement d’auto-administration. En revanche, lorsque la cocaïne est substituée par l’atomoxétine, les rats perdent peu à peu l’intérêt pour le levier précédemment associé à l’auto-administration de cocaïne (Wee et Woolverton, 2004
).
Antidépresseurs
Il existe différentes classes d’antidépresseurs et l’ensemble des données de la littérature relève quelques cas de dépendance avec des molécules ayant des structures chimiques de type amphétaminique (exemple de la tianeptine et de l’amineptine), et très peu de cas de mésusage et de pharmacodépendance chez les patients traités avec les autres classes d’antidépresseurs parmi lesquels des inhibiteurs de recapture de la sérotonine et/ou de la noradrénaline (Haddad, 1999
). Ces observations sont confortées par les études neurobiologiques.
L’apparition d’une pharmacodépendance pourrait être en lien avec la rupture d’une régulation mutuelle entre les neurones noradrénergiques et sérotoninergiques (revue dans Tassin, 2008
). Il a en effet été proposé l’hypothèse d’une régulation réciproque entre les neurones noradrénergiques et sérotoninergiques, par l’intermédiaire respectivement des récepteurs 5-HT2A et α1b-adrénergiques (figure 12.3
). Dans le modèle de sensibilisation comportementale, il apparaît que le couplage entre ces deux ensembles neuronaux disparaît lors de la répétition d’injection de drogues (licites ou illicites). De façon très intéressante, cette perte de régulation entre les systèmes noradrénergique et sérotoninergique n’est pas observée avec les antidépresseurs, en particulier la clomipramine et la venlafaxine, qui sont deux molécules capables d’inhiber à la fois les systèmes de recapture de la sérotonine et de la noradrénaline. L’étude de ce découplage spécifique pourrait donc représenter un bon marqueur pour différencier les produits avec un potentiel addictif (Tassin, 2008
).
Si le risque de pharmacodépendance aux antidépresseurs semble faible dans la population générale, il peut néanmoins être plus important chez des patients ayant rencontré au préalable des problèmes de dépendance à d’autres drogues ou à l’alcool (Quaglio et coll., 2008
). Cette plus grande vulnérabilité peut avoir plusieurs origines. Il est en effet bien connu que les drogues sont capables de modifier les réseaux neuronaux (connexions entre les différentes régions cérébrales), l’homéostasie, ou encore induire des modifications épigénétiques, à l’origine de différentes modulations transcriptomiques. Par ailleurs, une sensibilisation des systèmes sérotoninergique et noradrénergique apparaît de façon systématique après une exposition prolongée aux substances psychotropes (cocaïne, morphine, alcool) chez les rongeurs qui se traduit en une libération extracellulaire très importante de ces neurotransmetteurs lors de la réinjection de la drogue (Tassin, 2008
; Lanteri et coll., 2008
). Sur ces cerveaux « modifiés », les actions des antidépresseurs sur les systèmes de neurotransmission (notamment sérotonine, noradrénaline, dopamine) pourraient alors être amplifiées, ceci pouvant expliquer les risques plus élevés de pharmacodépendance.
Pour résumer, même si certains médicaments psychotropes partagent des cibles communes avec la cocaïne, les données actuelles indiquent des risques de pharmacodépendance relativement faibles, mais avec un impact sans doute non négligeable en regard de leur utilisation très large. Ceci peut s’expliquer par des propriétés pharmacodynamiques différentes, en particulier la sélectivité vis-à-vis des cibles. Les propriétés pharmacocinétiques doivent également être prises en compte. En effet, certaines molécules peuvent être substrats de transporteurs d’influx ou d’efflux, aussi bien au niveau de la barrière hémato-encéphalique que des membranes neuronales, pouvant ainsi modifier les concentrations cérébrales et la distribution intra-neuronale
versus espace extracellulaire, à l’origine de différences dans les réponses pharmacologiques, comme par exemple le développement ou non d’une pharmacodépendance (Amphoux et coll., 2006
; Dutheil et coll., 2010
).
Pharmacodépendance aux opioïdes dans le cadre d’un traitement
Les opioïdes sont prescrits pour la prise en charge d’une douleur ou dans le cadre d’un traitement de substitution aux opiacés. Les préparations pharmaceutiques à base d’opioïdes sont nombreuses dans la pharmacopée, avec des ligands opioïdes ayant des propriétés pharmacologiques différentes. Les opioïdes sont classés en fonction de leur affinité vis-à-vis des quatre types connus de récepteurs aux opiacés :
mu, delta, kappa et le récepteur de la nociceptine/orphanine
4
Peptide endogène exerçant un effet algésique
appelé ORL1
(opioid receptor like-1).
Traitement de la douleur
Parmi la diversité des traitements antalgiques, médicamenteux ou autres, les analgésiques opioïdes sont les plus puissants et, à ce titre, ce sont des médicaments majeurs en clinique dans le traitement des douleurs intenses aiguës ou chroniques. La France avait pris un grand retard dans le domaine de leur emploi. La mise en place des différents plans de lutte contre la douleur
5
Un premier plan de lutte contre la douleur a été mis en place en 1998-2000, suivi d’un deuxième programme (2002-2005) et d’un plan d’amélioration de la prise en charge de la douleur (2006-2010). http://www.sante.gouv.fr/plans-de-lutte-contre-la-douleur.html
par les autorités sanitaires de notre pays a permis, d’une part, une utilisation plus appropriée des analgésiques opioïdes par le monde médical et, d’autre part, une meilleure perception de ceux-ci par les patients.
Seuls les agonistes pouvant activer les récepteurs
mu sont capables de produire des réponses analgésiques fortes, mais ce sont également ceux utilisés dans le cadre d’usage abusif (voir la démonstration avec l’oxycodone dans Beardsley et coll., 2004
). Le développement d’une tolérance (nécessitant une augmentation des doses) et la crainte d’abus et de dépendance sont les facteurs majeurs limitant l’accès aux analgésiques opioïdes (Fields, 2011
). En pratique, pour la plupart des patients, l’usage médical rationnel des opioïdes pour soulager la douleur n’entraîne pas ou peu de mésusage et de comportement addictif. Ces observations sont également à rapprocher d’études précliniques qui montrent une diminution du potentiel renforçant de la morphine mesuré dans le test de préférence de place conditionnée chez des animaux présentant une douleur (Betourne et coll., 2008
). Néanmoins, une pharmacodépendance peut apparaître dans certains cas, en particulier quand les patients continuent à consommer des opioïdes même s’ils ne ressentent plus de douleur. Ces consommations prolongées peuvent conduire à des altérations morphologiques et fonctionnelles de certaines régions cérébrales (Upadhyay et coll., 2010
), comme l’amygdale qui est une région connue pour jouer un rôle dans les addictions (Koob et Volkow, 2010
).
En fonction de leurs propriétés pharmacologiques spécifiques, tous les agonistes opioïdes capables de se fixer sur les récepteurs
mu n’auront pas le même potentiel d’induction d’une pharmacodépendance lorsqu’ils sont utilisés dans un contexte d’addiction. Par exemple, le tramadol, qui est un agoniste opioïde atypique, avec des propriétés d’agoniste partiel (opioïde faible, niveau antalgique 2 de l’OMS)
6
L’OMS a proposé une classification à trois niveaux pour l’utilisation des antalgiques. Elle est essentiellement basée sur l’intensité de la douleur. Niveau 1 : les antalgiques à action préférentiellement « périphérique » pour une douleur légère à modérée. Niveau 2 : les antalgiques opiacés faibles pour une douleur modérée à sévère et/ou en cas d’échec des antalgiques de niveau 1. Niveau 3 : les antalgiques opiacés forts pour une douleur sévère et/ou en cas d’échec des antalgiques de niveau 2.
, présente un faible pouvoir addictif par comparaison à d’autres agonistes opioïdes comme la morphine et le rémifentanil, qui sont des agonistes opioïdes
mu forts (niveau antalgique 3 de l’OMS) (O’Connor et Mead, 2010
). La démonstration a été faite en comparant les résultats obtenus chez différentes espèces animales, et en utilisant différents tests comportementaux (Epstein et coll., 2006
).
Traitements de substitution aux opiacés
L’action d’un traitement de substitution vise, en prévenant la symptomatologie psychique et physique du manque, à stabiliser la consommation d’opiacés (principalement l’héroïne) ou, pour le moins, à la diminuer et insérer le patient dans une logique de soins. Ce traitement consiste donc à remplacer la consommation d’héroïne par la prise de médicaments opiacés (méthadone ou buprénorphine haut dosage) par une voie qui évite les effets de pic (voie orale pour la méthadone ou voie sublinguale pour la buprénorphine). Des essais cliniques sont également en cours aux États-Unis avec des implants sous-cutanés de buprénorphine qui permettent de délivrer une dose faible et régulière de buprénorphine à partir d’une dose unique implantée pour 6 mois (Ling et coll., 2010
).
L’usage détourné par injection de la méthadone, telle qu’elle est disponible en France (voie orale), est très difficile. Ceci tient au fait de la présentation galénique de la méthadone sous forme soit de sirop, soit de gélules contenant un gélifiant, rendant difficile, voire impossible la solubilisation du contenu et son transfert dans une seringue.
En revanche, le mésusage (et le détournement) de la buprénorphine demeure fréquent, certains patients n’hésitant pas à cumuler les prescriptions et à s’injecter un filtrat de comprimés. Dans le but d’éviter ces mésusages par injection, la buprénorphine a été associée à un antagoniste opiacé, la naloxone. L’intérêt de cette association est lié à la très mauvaise biodisponibilité (fraction qui atteint la circulation sanguine) de la naloxone par voie orale, alors qu’elle est par définition de 100 % par voie intraveineuse. Autrement dit, cet antagoniste est plus performant par voie intraveineuse que par voie orale. Ainsi, du fait de sa grande biodisponibilité par voie intraveineuse, la naloxone bloquera les effets de la buprénorphine si le médicament buprénorphine/naloxone est injecté (à l’inverse d’une prise par voie orale). Les résultats cliniques montrent globalement que cette association buprénorphine/naloxone n’empêche pas le mésusage par injection, mais le potentiel addictif semble un peu plus faible que celui de la buprénorphine seule (Mammen et Bell, 2009
; Comer et coll., 2010
).
Pharmacodépendance aux benzodiazépines et substances apparentées
Parmi les différents médicaments psychotropes disponibles sur le marché, les membres de la classe des benzodiazépines sont largement prescrits en France et, pour certains, figurent parmi les plus à risque d’engendrer une dépendance.
Benzodiazépines, substances apparentées, et récepteurs au GABA
Le récepteur GABAA présente, en dehors du site de liaison du GABA, une variété d’autres sites récepteurs topographiquement distincts et capables de reconnaître des substances actives, comme les benzodiazépines ou des hypnotiques (zolpidem ou zopiclone par exemple, qui ont des structures chimiques différentes de celles des benzodiazépines, mais qui agissent sur le même site et qui sont souvent par extension nommés comme benzodiazépines). Ces substances agissent de manière allostérique avec les sites récepteurs du GABA et modulent la réponse GABAergique. En effet, les benzodiazépines et molécules apparentées ne stimulent pas directement le récepteur mais le rendent plus efficace en augmentant la fréquence d’ouverture du canal chlore lorsque le GABA se fixe. Les récepteurs GABAA sont des glycoprotéines transmembranaires, qui présentent une grande hétérogénéité de structure mais également une hétérogénéité de réponse pharmacologique, dont les conséquences sont encore mal connues. En effet, ils sont constitués de cinq sous-unités (alpha, bêta, gamma et delta, thêta), chacune de ces sous-unités présentant différents sous-types : six types de sous-unités alpha, trois de sous-unités bêta, trois de sous-unités gamma et un type de sous-unités delta.
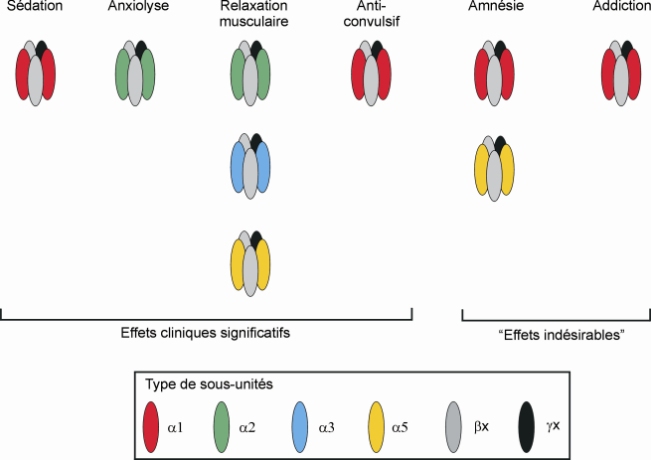
Les sous-unités
alpha ont fait l’objet d’études plus approfondies. Il a ainsi pu être montré en utilisant des souris génétiquement modifiées que les sous-unités
alpha2 et
alpha3 sont impliquées dans les effets anxiolytiques et myorelaxants, et les sous-unités
alpha5 dans l’apprentissage et la mémoire (figure 12.7
) (Atack, 2011a
et 2011b
). Par ailleurs, l’activation de la sous-unité
alpha1 est suffisante (mais pas nécessaire) pour observer une auto-administration de benzodiazépines, alors que l’activation des sous-unités
alpha2, 3 et/ou 5 est suffisante pour induire ce comportement, mais de façon beaucoup moins importante que la sous-unité
alpha1 (Rowlett et coll., 2005
; Licata et coll., 2011
).
Chaque benzodiazépine a un profil pharmacologique spécifique, présentant en particulier des affinités différentes pour les récepteurs GABA
A en fonction de leur composition (Hanson et coll., 2008
). Les benzodiazépines et substances apparentées ayant une forte affinité pour les sous-unités
alpha1 (exemple du zolpidem) présentent un potentiel addictif plus élevé chez l’animal (Tan et coll., 2010
), et aussi un effet de «
high » en clinique (Licata et coll., 2011
). Il a en particulier été montré dans le test de discrimination chez le singe, le maintien d’un comportement d’auto-administration quand la cocaïne est substituée par le zolpidem (Griffiths et coll., 1992
).
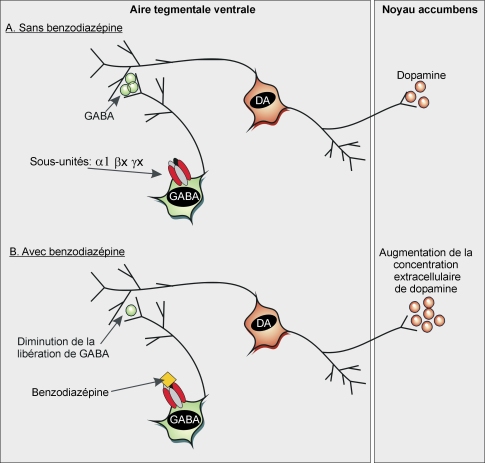
Activation des neurones dopaminergiques par les benzodiazépines
Comme évoqué au début de ce chapitre, les neurones dopaminergiques du système mésolimbique sont sous le contrôle inhibiteur de neurones GABAergiques présents dans l’aire tegmentale ventrale. Une étude récente a montré la présence de récepteurs GABA
A possédant spécifiquement des sous-unités
alpha1 sur ces interneurones GABAergiques (Tan et coll., 2010
et 2011
). L’activation de ces récepteurs par les benzodiazépines ayant une bonne affinité pour les sous-unités
alpha1 (comme le zolpidem) conduit à une entrée d’ions Cl
− dans le neurone, induisant une hyperpolarisation de celui-ci. Il y a donc inhibition du neurone GABAergique et diminution de la libération de GABA, qui ne peut plus exercer son rôle de frein sur le neurone dopaminergique (Tan et coll., 2010
et 2011
). La conséquence est une augmentation de libération de dopamine dans le noyau accumbens (figure 12.8
). Ce mécanisme est tout à fait comparable à celui observé avec les opioïdes et les cannabinoïdes ; ils activent aussi leurs récepteurs respectifs présents sur les neurones GABAergiques de l’aire tegmentale ventrale qui contrôlent l’activité des neurones dopaminergiques du système mésocortolimbique.
Sevrage aux benzodiazépines
Les récepteurs ionotropes AMPA et NMDA, activés par le glutamate, jouent un rôle important dans les mécanismes neurobiologiques des addictions. Comme les récepteurs GABAA, ces récepteurs au glutamate sont composés de différentes sous-unités, et en fonction de la nature de ces sous-unités, la fonctionnalité des récepteurs peut être modifiée. Les récepteurs NMDA sont des hétérotétramères, formés de deux sous-unités NR1 et de deux sous-unités NR2. Les sous-unités NR2 (NR2A, NR2B, NR2C, NR2D) spécifient les propriétés électrophysiologiques des récepteurs NMDA, telle leur sensibilité au glutamate, leur perméabilité au calcium, ou l’inhibition du magnésium. Les récepteurs AMPA sont également des hétérotétramères résultant de l’association de deux sous-unités différentes parmi les quatre existantes, nommées GluR1, GluR2, GluR3 et GluR4. Une fois le récepteur activé, il y a ouverture d’un canal permettant la sortie de potassium de la cellule, et l’entrée de sodium et de calcium. Cette entrée de calcium est dépendante des sous-unités GluR2, leur présence rendant le canal imperméable au calcium. Suite à ces entrées d’ions, il y a dépolarisation de la membrane du neurone cible du glutamate.
Plusieurs études démontrent le rôle important des récepteurs NMDA et AMPA dans les phénomènes de plasticité synaptique, et en particulier dans les mécanismes de pharmacodépendance. Il a été montré que la sensibilisation comportementale à la cocaïne est associée à une modification, à la surface membranaire, des sous-unités des récepteurs AMPA dans le noyau accumbens, avec en particulier une augmentation du rapport GluR1/GluR2 (davantage de GluR1 et moins de GluR2). Les mêmes phénomènes sont observés lors d’un sevrage aux benzodiazépines chez les rongeurs, avec une hypersensibilité des récepteurs AMPA suite au recrutement de sous-unités GluR1 à la place de sous-unités GluR2, ce qui augmente la perméabilité au calcium et induit une dépolarisation de la membrane plus importante (Song et coll., 2007
; Shen et coll., 2010
). Parallèlement à ce phénomène, la composition des récepteurs NMDA est aussi modifiée, avec une diminution des sous-unités NR2B, rendant le canal moins perméable aux ions, et qui aurait pour fonction de s’opposer à l’hyperexcitabilité des récepteurs AMPA, limitant ainsi les effets anxiogéniques du sevrage (Das et coll., 2010
; Shen et Tietz, 2011
).
Pharmacodépendance aux anesthésiques
De structure très proche de la phencyclidine (PCP)
7
Ancien anesthésique des années 1950 retiré du marché
, la kétamine (Kétalar® à usage humain et vétérinaire) et la tilétamine (Zoletil® à usage vétérinaire) sont des anesthésiques généraux non-barbituriques qui sont toutes deux classées comme stupéfiants en France. Ce sont des antagonistes des récepteurs glutamatergiques de type NMDA. Elles dépriment certaines régions cérébrales comme le thalamus et le cortex, tandis que d’autres structures, notamment concernant le système limbique, restent activées.
La kétamine et la tilétamine exercent des effets hallucinogènes puissants et leur usage détourné est principalement rencontré en milieu festif, surtout chez les jeunes présentant le même profil que les consommateurs d’ecstasy. Ces substances modifient la perception auditive, visuelle, mais également du temps et de l’espace. Elles sont très prisées pour la sensation de décorporation qu’elles engendrent, mais également pour gérer la « descente » associée aux produits stimulants. Dans les deux cas, le risque de pharmacodépendance est élevé.
Anesthésique général d’action rapide et brève, le propofol (Diprivan®) possède une activité antagoniste des récepteurs NMDA qui contribuerait aux effets anesthésiques et amnésiques. Il agit également sur les récepteurs GABAA à l’origine d’une dépression de l’activité cérébrale.
Le potentiel d’abus et de dépendance du propofol est étayé par des données très complètes sur le plan pharmacologique
in vivo chez l’animal et chez l’Homme. Toutefois, il est intéressant de noter qu’il y a une grande disparité de réponse dans un modèle d’auto-administration chez le rat, certains animaux ne développant pas de comportement d’auto-administration. Cette même diversité a été également rapportée dans des études cliniques (Zacny et coll., 1993
).
Interactions médicamenteuses
Les avancées de la recherche ont permis d’identifier un certain nombre de cibles pharmacologiques qui jouent un rôle dans les différentes étapes conduisant à un comportement d’addiction.
Interactions opioïdes-benzodiazépines
Les récepteurs opioïdes
mu et les récepteurs GABA
A (possédant les sous-unités
alpha1) sont co-localisés sur les interneurones GABAergiques dans l’aire tegmentale ventrale. Ces deux récepteurs
mu et GABA
A sont capables, après activation, de lever l’inhibition sur les neurones dopaminergiques qui atteignent les structures limbiques. Il n’est donc pas surprenant d’observer une potentialisation des effets renforçants de l’héroïne par les benzodiazépines dans le modèle de la préférence de place conditionnée (Walker et Ettenberg, 2001
).
Dans une étude où des rats sont exposés à la buprénorphine seule ou en association avec des benzodiazépines, il a été mis en évidence, dans certaines régions cérébrales (amygdale, cortex, hippocampe, hypothalamus et thalamus), des augmentations de densité des récepteurs
mu uniquement chez les rats exposés à l’association buprénorphine et benzodiazépine (Poisnel et coll., 2009
).
Par ailleurs, les cas référencés de décès chez les usagers de buprénorphine sont survenus dans le contexte d’un mauvais usage de ce médicament, par voie intraveineuse ou en association avec des benzodiazépines et de l’alcool (Pirnay et coll., 2004
).
Autres interactions potentielles
Les études cliniques et précliniques suggèrent l’existence d’interactions entre les systèmes dopaminergiques et muscariniques. Les antimuscariniques (ou atropiniques) réduisent l’hyperactivité cholinergique striatale résultant de la réduction du tonus inhibiteur dopaminergique observé dans la maladie de Parkinson. Utilisé comme antiparkinsonien, le trihexyphénidyle est un atropinique de synthèse commercialisé en France sous le nom d’Artane®. Dans cette classe de médicament, il semble être celui qui possède le potentiel d’abus et de dépendance le plus élevé. Il est souvent consommé en association avec d’autres drogues illicites, l’alcool ou des benzodiazépines (Frauger et coll., 2003
; Nappo et coll., 2005
). C’est un antagoniste des récepteurs muscariniques M1, et il est capable de potentialiser la libération de dopamine induite par la cocaïne, ainsi que ses effets comportementaux (Tanda et Katz, 2007
; Tanda et coll., 2007
).
Le topiramate (Epitomax®) est habituellement utilisé comme antiépileptique et en traitement préventif de la migraine. Il augmente la fréquence d’activation des récepteurs GABA
A par le GABA ainsi que la capacité de ce neuromédiateur à induire l’influx des ions chlore dans les neurones. Par ailleurs, le topiramate antagonise faiblement l’activité excitatrice du glutamate au niveau des récepteurs de type kaïnate/AMPA. Il n’a pas d’effet apparent au niveau des récepteurs de type NMDA. À l’heure actuelle, aucun cas de pharmacodépendance ne semble déclaré avec ce produit. Néanmoins, des études rapportent que le topiramate par voie orale (25 à 50 mg) chez l’Homme est capable d’augmenter les effets subjectifs de la nicotine injectée par voie intraveineuse (Sofuoglu et coll., 2006
). Une autre étude montre également une augmentation des effets subjectifs positifs de la méthamphétamine injectée par voie intraveineuse, après administration par voie orale de topiramate (100 à 200 mg) (Johnson et coll., 2007
).
La prégabaline (Lyrica®) est autorisée dans le traitement adjuvant des épilepsies partielles rebelles au traitement médicamenteux, le traitement de la douleur neurogène périphérique chez l’adulte, et les troubles anxieux. Il s’agit d’un analogue structural du GABA (3-isobutyl GABA), proche de la gabapentine. De par cette structure chimique proche du GABA, le risque d’abus peut exister. Effectivement depuis 2010, quelques indicateurs mettent en évidence un usage avec un potentiel d’abus de cette molécule (Filipetto et coll., 2010
; Schwan et coll., 2010
; Schifano et coll., 2011
).
Le modafinil est l’un des stimulants utilisés médicalement dans le traitement de la narcolepsie et de l’hypersomnie idiopathique. Les cibles pharmacologiques de cette molécule sont nombreuses. Parmi celles-ci, le système de recapture de la dopamine que le modafinil est capable d’inhiber, ce qui a pour conséquence d’augmenter la motivation pour la réalisation d’une tâche spécifique chez le rat (Young et Geyer, 2010
). Il a été montré qu’il augmente la concentration extracellulaire de dopamine dans le striatum chez le singe (Andersen et coll., 2010
). Le modafinil pourrait également agir sur d’autres systèmes. En particulier, cette molécule serait capable d’activer le système histaminique, via les neurones orexines (Ishizuka et coll., 2010
). Ce résultat est à mettre en perspective avec ce que l’on sait maintenant du système orexine dans les addictions (Aston-Jones et coll., 2010
). Ce neuropeptide semble jouer un rôle important dans les processus de rechute à la cocaïne. Malgré ces résultats précliniques très récents, les études cliniques plus anciennes ne semblaient pas mettre en évidence un mésusage du modafinil, qui aurait peu de propriétés discriminatives vis-à-vis de la cocaïne (Rush et coll., 2002
) et peu de propriétés euphorisantes (O’Brien et coll., 2006
).
En conclusion, les études cliniques et précliniques soulignent bien les risques de mésusage et de pharmacodépendance des médicaments psychotropes. Les avancées récentes sur les mécanismes neurobiologiques des dépendances aux drogues licites et illicites permettent de mieux appréhender ces risques. Néanmoins, il est important de noter que cette évaluation ne peut être pertinente que si la pharmacologie des médicaments est bien connue. En effet, un certain nombre de circuits neuronaux et de cibles pharmacologiques sont maintenant bien identifiés comme jouant un rôle essentiel dans la mise en place des conduites addictives et leur maintien. Toutes les molécules capables d’interagir sur ces circuits et/ou avec ces cibles peuvent donc présenter des risques de mésusages et de pharmacodépendance.
Même si les médicaments psychotropes sont capables d’agir sur les différentes cibles, il ne faut pas oublier que les propriétés pharmacocinétiques doivent également être intégrées dans l’équation finale de l’évaluation des risques de mésusages et de pharmacodépendance. L’utilisation contrôlée des médicaments psychotropes, en suivant les règles de prescription, ne conduira que très rarement à des pharmacodépendances. Le risque deviendra beaucoup plus élevé s’il y a mésusage (en particulier modification de la voie d’administration, avec injections par voie intraveineuse), favorisant des effets pic au niveau des cibles pharmacologiques cérébrales.
Des adaptations neurobiologiques responsables des pharmacodépendances peuvent également apparaître au cours de consommations prolongées avec certains médicaments. Il est donc important que pour ces médicaments les durées de traitement ne dépassent pas les durées de prescriptions maximales fixées par les agences sanitaires.
L’analyse de la littérature montre également que le risque de mésusage et de pharmacodépendance n’est pas identique chez tous les individus. Il y a bien sûr des facteurs génétiques qui peuvent influencer ce risque, mais également des facteurs épigénétiques et la trajectoire préalable de chacun vis-à-vis de la consommation de composés psychotropes. En effet, des histoires d’usages abusifs ou de mésusages de ces composés peuvent induire des altérations neurobiologiques plus ou moins persistantes, rendant certains patients/utilisateurs plus vulnérables à une exposition à un médicament psychotrope.
Bibliographie
[1] AMPHOUX A, VIALOU V, DRESCHER E, BRUSS M, MANNOURY LA COUR C, et coll. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain.
Neuropharmacology. 2006;
50:941
-952

[2] ANDERSEN ML, KESSLER E, MURNANE KS, MCCLUNG JC, TUFIK S, HOWELL LL. Dopamine transporter-related effects of modafinil in rhesus monkeys.
Psychopharmacology (Berl). 2010;
210:439
-448
[3] ASTON-JONES G, SMITH RJ, SARTOR GC, MOORMAN DE, MASSI L, et coll. Lateral hypothalamic orexin/hypocretin neurons: A role in reward-seeking and addiction.
Brain Res. 2010;
1314:74
-90
[4] ATACK JR. GABA(A) Receptor Subtype-Selective Modulators. I. alpha2/alpha3-Selective Agonists as Non-Sedating Anxiolytics.
Curr Top Med Chem. 2011a;
11:1176
-1202
[5] ATACK JR. GABA(A) Receptor Subtype-Selective Modulators. II. alpha5-Selective Inverse Agonists for Cognition Enhancement.
Curr Top Med Chem. 2011b;
11:1203
-1214
[6] ATOR NA, GRIFFITHS RR. Principles of drug abuse liability assessment in laboratory animals.
Drug Alcohol Depend. 2003;
70:S55
-S72
[7] BALSTER RL, BIGELOW GE. Guidelines and methodological reviews concerning drug abuse liability assessment.
Drug Alcohol Depend. 2003;
70:S13
-S40
[8] BEARDSLEY PM, ACETO MD, COOK CD, BOWMAN ER, NEWMAN JL, et coll. Discriminative stimulus, reinforcing, physical dependence, and antinociceptive effects of oxycodone in mice, rats, and rhesus monkeys.
Exp Clin Psychopharmacol. 2004;
12:163
-172
[9] BERMAN SM, KUCZENSKI R, MCCRACKEN JT, LONDON ED. Potential adverse effects of amphetamine treatment on brain and behavior: a review.
Mol Psychiatry. 2009;
14:123
-142
[10] BETOURNE A, FAMILIADES J, LACASSAGNE L, HALLEY H, CAZALES M, et coll. Decreased motivational properties of morphine in mouse models of cancerous- or inflammatory-chronic pain: implication of supraspinal neuropeptide FF(2) receptors.
Neuroscience. 2008;
157:12
-21
[11] BOTLY LC, BURTON CL, RIZOS Z, FLETCHER PJ. Characterization of methylphenidate self-administration and reinstatement in the rat.
Psychopharmacology (Berl). 2008;
199:55
-66
[12] BYMASTER FP, KATNER JS, NELSON DL, HEMRICK-LUECKE SK, THRELKELD PG, et coll. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder.
Neuropsychopharmacology. 2002;
27:699
-711
[13] COMER SD, ASHWORTH JB, SULLIVAN MA, VOSBURG SK, SACCONE PA, et coll. Relationship between rate of infusion and reinforcing strength of oxycodone in humans.
J Opioid Manag. 2009;
5:203
-212
[14] COMER SD, SULLIVAN MA, VOSBURG SK, MANUBAY J, AMASS L, et coll. Abuse liability of intravenous buprenorphine/naloxone and buprenorphine alone in buprenorphine-maintained intravenous heroin abusers.
Addiction. 2010;
105:709
-718
[15] CONE EJ. Pharmacokinetics and pharmacodynamics of cocaine.
J Anal Toxicol. 1995;
19:459
-478
[16] DAS P, ZERDA R, ALVAREZ FJ, TIETZ EI. Immunogold electron microscopic evidence of differential regulation of GluN1, GluN2A, and GluN2B, NMDA-type glutamate receptor subunits in rat hippocampal CA1 synapses during benzodiazepine withdrawal.
J Comp Neurol. 2010;
518:4311
-4328
[17] DUTHEIL F, JACOB A, DAUCHY S, BEAUNE P, SCHERRMANN JM, et coll. ABC transporters and cytochromes P450 in the human central nervous system: influence on brain pharmacokinetics and contribution to neurodegenerative disorders.
Expert Opin Drug Metab Toxicol. 2010;
6:1161
-1174
[18] EPSTEIN DH, PRESTON KL, JASINSKI DR. Abuse liability, behavioral pharmacology, and physical-dependence potential of opioids in humans and laboratory animals: lessons from tramadol.
Biol Psychol. 2006;
73:90
-99
[19] FERGUSON SA, BOCTOR SY. Cocaine responsiveness or anhedonia in rats treated with methylphenidate during adolescence.
Neurotoxicol Teratol. 2010;
32:432
-442
[20] FIELDS HL. The Doctor’s Dilemma: Opiate Analgesics and Chronic Pain.
Neuron. 2011;
69:591
-594
[21] FILIPETTO FA, ZIPP CP, COREN JS. Potential for pregabalin abuse or diversion after past drug-seeking behavior.
J Am Osteopath Assoc. 2010;
110:605
-607
[22] FRAUGER E, THIRION X, CHANUT C, NATALI F, DEBRUYNE D, et coll. Misuse of trihexyphenidyl (Artane, Parkinane): recent trends.
Therapie. 2003;
58:541
-547
[23] GASIOR M, BERGMAN J, KALLMAN MJ, PARONIS CA. Evaluation of the reinforcing effects of monoamine reuptake inhibitors under a concurrent schedule of food and i.v. drug delivery in rhesus monkeys.
Neuropsychopharmacology. 2005;
30:758
-764
[24] GRIFFITHS RR, SANNERUD CA, ATOR NA, BRADY JV. Zolpidem behavioral pharmacology in baboons: self-injection, discrimination, tolerance and withdrawal.
J Pharmacol Exp Ther. 1992;
260:1199
-1208
[25] HADDAD P. Do antidepressants have any potential to cause addiction?.
J Psychopharmacol. 1999;
13:300
-307
[26] HANSON SM, MORLOCK EV, SATYSHUR KA, CZAJKOWSKI C. Structural requirements for eszopiclone and zolpidem binding to the gamma-aminobutyric acid type-A (GABAA) receptor are different.
J Med Chem. 2008;
51:7243
-7252
[27] HYMAN SE, MALENKA RC, NESTLER EJ. Neural mechanisms of addiction: the role of reward-related learning and memory.
Annu Rev Neurosci. 2006;
29:565
-598
[28] ISHIZUKA T, MUROTANI T, YAMATODANI A. Modanifil activates the histaminergic system through the orexinergic neurons.
Neurosci Lett. 2010;
483:193
-196
[29] JOHNSON BA, ROACHE JD, AIT-DAOUD N, WELLS LT, WALLACE CL, et coll. Effects of acute topiramate dosing on methamphetamine-induced subjective mood.
Int J Neuropsychopharmacol. 2007;
10:85
-98
[30] KATZUNG B, MASTERS S, TREVOR A.Basic and Clinical Pharmacology, 11th Edition. McGraw-Hill Companies. 2009;
[31] KIM Y, TEYLAN MA, BARON M, SANDS A, NAIRN AC, et coll. Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens.
Proc Natl Acad Sci USA. 2009;
106:2915
-2920
[32] KOOB GF, VOLKOW ND. Neurocircuitry of addiction.
Neuropsychopharmacology. 2010;
35:217
-238
[33] LANTERI C, SALOMON L, TORRENS Y, GLOWINSKI J, TASSIN JP. Drugs of abuse specifically sensitize noradrenergic and serotonergic neurons via a non-dopaminergic mechanism.
Neuropsychopharmacology. 2008;
33:1724
-1734
[34] LICATA SC, MASHHOON Y, MACLEAN RR, LUKAS SE. Modest abuse-related subjective effects of zolpidem in drug-naive volunteers.
Behav Pharmacol. 2011;
22:160
-166
[35] LING W, CASADONTE P, BIGELOW G, KAMPMAN KM, PATKAR A, et coll. Buprenorphine implants for treatment of opioid dependence: a randomized controlled trial.
JAMA. 2010;
304:1576
-1583
[36] MAMMEN K, BELL J. The clinical efficacy and abuse potential of combination buprenorphine-naloxone in the treatment of opioid dependence.
Expert Opin Pharmacother. 2009;
10:2537
-2544
[37] MCGOVERN RW, MIDDAUGH LD, PATRICK KS, GRIFFIN WC, III. The discriminative stimulus properties of methylphenidate in C57BL/6J mice.
Behav Pharmacol. 2011;
22:14
-22
[38] MERIRINNE E, KANKAANPAA A, SEPPALA T. Rewarding properties of methylphenidate: sensitization by prior exposure to the drug and effects of dopamine D1- and D2-receptor antagonists.
J Pharmacol Exp Ther. 2001;
298:539
-550
[39] NAPPO SA, DE OLIVEIRA LG, SANCHEZ ZM, CARLINI EDE A. Trihexyphenidyl (Artane): a Brazilian study of its abuse.
Subst Use Misuse. 2005;
40:473
-482
[40] O’BRIEN CP, DACKIS CA, KAMPMAN K. Does modafinil produce euphoria?.
Am J Psychiatry. 2006;
163:1109
[41] O’CONNOR EC, MEAD AN. Tramadol acts as a weak reinforcer in the rat self-administration model, consistent with its low abuse liability in humans.
Pharmacol Biochem Behav. 2010;
96:279
-286
[42] PIRNAY S, BORRON SW, GIUDICELLI CP, TOURNEAU J, BAUD FJ, RICORDEL I. A critical review of the causes of death among post-mortem toxicological investigations: analysis of 34 buprenorphine-associated and 35 methadone-associated deaths.
Addiction. 2004;
99:978
-988
[43] POISNEL G, DHILLY M, LE BOISSELIER R, BARRE L, DEBRUYNE D. Comparison of five benzodiazepine-receptor agonists on buprenorphine-induced mu-opioid receptor regulation.
J Pharmacol Sci. 2009;
110:36
-46
[44] QUAGLIO G, SCHIFANO F, LUGOBONI F. Venlafaxine dependence in a patient with a history of alcohol and amineptine misuse.
Addiction. 2008;
103:1572
-1574
[45] ROWLETT JK, PLATT DM, LELAS S, ATACK JR, DAWSON GR. Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates.
Proc Natl Acad Sci USA. 2005;
102:915
-920
[46] RUSH CR, KELLY TH, HAYS LR, WOOTEN AF. Discriminative-stimulus effects of modafinil in cocaine-trained humans.
Drug Alcohol Depend. 2002;
67:311
-322
[47] SAMAHA AN, ROBINSON TE. Why does the rapid delivery of drugs to the brain promote addiction?.
Trends Pharmacol Sci. 2005;
26:82
-87
[48] SAMAHA AN, MALLET N, FERGUSON SM, GONON F, ROBINSON TE. The rate of cocaine administration alters gene regulation and behavioral plasticity: implications for addiction.
J Neurosci. 2004;
24:6362
-6370
[49] SCHENK S, IZENWASSER S. Pretreatment with methylphenidate sensitizes rats to the reinforcing effects of cocaine.
Pharmacol Biochem Behav. 2002;
72:651
-657
[50] SCHIFANO F, D’OFFIZI S, PICCIONE M, CORAZZA O, DELUCA P, et coll. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data.
Psychother Psychosom. 2011;
80:118122. Epub 2011 Jan 4.
[51] SCHINDLER CW, PANLILIO LV, THORNDIKE EB. Effect of rate of delivery of intravenous cocaine on self-administration in rats.
Pharmacol Biochem Behav. 2009;
93:375
-381
[52] SCHWAN S, SUNDSTRÖM A, STJERNBERG E, HALLBERG E, HALLBERG P. A signal for an abuse liability for pregabalin--results from the Swedish spontaneous adverse drug reaction reporting system.
Eur J Clin Pharmacol. 2010;
66:947953. Epub 2010 Jun 19.
[53] SELLINGS LH, MCQUADE LE, CLARKE PB. Characterization of dopamine-dependent rewarding and locomotor stimulant effects of intravenously-administered methylphenidate in rats.
Neuroscience. 2006;
141:1457
-1468
[54] SHEN G, TIETZ EI. Down-regulation of synaptic GluN2B subunit-containing N-methyl-D-aspartate receptors: a physiological brake on CA1 neuron alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid hyperexcitability during benzodiazepine withdrawal.
J Pharmacol Exp Ther. 2011;
336:265
-273
[55] SHEN G, VAN SICKLE BJ, TIETZ EI. Calcium/calmodulin-dependent protein kinase II mediates hippocampal glutamatergic plasticity during benzodiazepine withdrawal.
Neuropsychopharmacology. 2010;
35:1897
-1909
[56] SOETERS HS, HOWELLS FM, RUSSELL VA. Methylphenidate does not increase ethanol consumption in a rat model for attention-deficit hyperactivity disorder-the spontaneously hypertensive rat.
Metab Brain Dis. 2008;
23:303
-314
[57] SOFUOGLU M, POLING J, MOURATIDIS M, KOSTEN T. Effects of topiramate in combination with intravenous nicotine in overnight abstinent smokers.
Psychopharmacology (Berl). 2006;
184:645
-651
[58] SONG J, SHEN G, GREENFIELD LJ, JR., TIETZ EI. Benzodiazepine withdrawal-induced glutamatergic plasticity involves up-regulation of GluR1-containing alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in Hippocampal CA1 neurons.
J Pharmacol Exp Ther. 2007;
322:569
-581
[59] STEINER H, VAN WAES V, MARINELLI M. Fluoxetine potentiates methylphenidate-induced gene regulation in addiction-related brain regions: concerns for use of cognitive enhancers?.
Biol Psychiatry. 2010;
67:592
-594
[60] SUSSMAN S, PENTZ MA, SPRUIJT-METZ D, MILLER T. Misuse of ″study drugs″: prevalence, consequences, and implications for policy.
Subst Abuse Treat Prev Policy. 2006;
1:15
[61] SUZUKI T, SHINDO K, MIYATAKE M, KUROKAWA K, HIGASHIYAMA K, et coll. Lack of development of behavioral sensitization to methylphenidate in mice: correlation with reversible astrocytic activation.
Eur J Pharmacol. 2007;
574:39
-48
[62] SVETLOV SI, KOBEISSY FH, GOLD MS. Performance enhancing, non-prescription use of Ritalin: a comparison with amphetamines and cocaine.
J Addict Dis. 2007;
26:1
-6
[63] SWANSON JM, VOLKOW ND. Serum and brain concentrations of methylphenidate: implications for use and abuse.
Neurosci Biobehav Rev. 2003;
27:615
-621
[64] TAN KR, BROWN M, LABOUEBE G, YVON C, CRETON C, et coll. Neural bases for addictive properties of benzodiazepines.
Nature. 2010;
463:769
-774
[65] TAN KR, RUDOLPH U, LUSCHER C. Hooked on benzodiazepines: GABAA receptor subtypes and addiction.
Trends Neurosci. 2011;
34:188
-197
[66] TANDA G, KATZ JL. Muscarinic preferential M(1) receptor antagonists enhance the discriminative-stimulus effects of cocaine in rats.
Pharmacol Biochem Behav. 2007;
87:400
-404
[67] TANDA G, EBBS AL, KOPAJTIC TA, ELIAS LM, CAMPBELL BL, et coll.Effects of muscarinic M1 receptor blockade on cocaine-induced elevations of brain dopamine levels and locomotor behavior in rats. J Pharmacol Exp Ther. 2007;
321:334344
[68] TASSIN JP. Uncoupling between noradrenergic and serotonergic neurons as a molecular basis of stable changes in behavior induced by repeated drugs of abuse.
Biochem Pharmacol. 2008;
75:85
-97
[69] THANOS PK, MICHAELIDES M, BENVENISTE H, WANG GJ, VOLKOW ND. Effects of chronic oral methylphenidate on cocaine self-administration and striatal dopamine D2 receptors in rodents.
Pharmacol Biochem Behav. 2007;
87:426
-433
[70] UPADHYAY J, MALEKI N, POTTER J, ELMAN I, RUDRAUF D, et coll. Alterations in brain structure and functional connectivity in prescription opioid-dependent patients.
Brain. 2010;
133:2098
-2114
[71] VAN WAES V, BEVERLEY J, MARINELLI M, STEINER H.Selective serotonin reuptake inhibitor antidepressants potentiate methylphenidate (Ritalin)-induced gene regulation in the adolescent striatum. Eur J Neurosci. 2010;
32:435447
[72] VOLKOW ND, SWANSON JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD.
Am J Psychiatry. 2003;
160:1909
-1918
[73] VOLKOW ND, WANG GJ, FOWLER JS, LOGAN J, GATLEY SJ, et coll. Reinforcing effects of psychostimulants in humans are associated with increases in brain dopamine and occupancy of D(2) receptors.
J Pharmacol Exp Ther. 1999;
291:409
-415
[74] WALKER BM, ETTENBERG A. Benzodiazepine modulation of opiate reward.
Exp Clin Psychopharmacol. 2001;
9:191
-197
[75] WEE S, WOOLVERTON WL. Evaluation of the reinforcing effects of atomoxetine in monkeys: comparison to methylphenidate and desipramine.
Drug Alcohol Depend. 2004;
75:271
-276
[76] WOOLVERTON WL, WANG Z. Relationship between injection duration, transporter occupancy and reinforcing strength of cocaine.
Eur J Pharmacol. 2004;
486:251
-257
[77] WOOTERS TE, NEUGEBAUER NM, RUSH CR, BARDO MT. Methylphenidate enhances the abuse-related behavioral effects of nicotine in rats: intravenous self-administration, drug discrimination, and locomotor cross-sensitization.
Neuropsychopharmacology. 2008;
33:11371148. Epub 2007 Jun 20.
[78] YANO M, STEINER H. Methylphenidate (Ritalin) induces Homer 1a and zif 268 expression in specific corticostriatal circuits.
Neuroscience. 2005;
132:855
-865
[79] YANO M, STEINER H. Methylphenidate and cocaine: the same effects on gene regulation?.
Trends Pharmacol Sci. 2007;
28:588
-596
[80] YOUNG JW, GEYER MA. Action of modafinil--increased motivation via the dopamine transporter inhibition and D1 receptors?.
Biol Psychiatry. 2010;
67:784
-787
[81] ZACNY JP, LICHTOR JL, ZARAGOZA JG, COALSON DW, UITVLUGT A, et coll. Assessing the behavioral effects and abuse potential of propofol bolus injections in healthy volunteers.
Drug Alcohol Depend. 1993;
32:45
-57
→ Aller vers SYNTHESE