Connaissances fondamentales
2007
| ANALYSE |
3-
Dégénérescences neurofibrillaires et protéines Tau
Tau (Tubulin-Associated Unit) est une protéine neuronale associée aux microtubules. Elle a été initialement décrite comme le « facteur Tau », un élément capable d'induire la polymérisation de la tubuline en microtubules (Cleveland et coll., 1977 ). En réalité, elle favorise la polymérisation et la stabilité des microtubules. Depuis 1985, elle est aussi identifiée comme le composant majeur des paires de filaments en hélice (Paired Helical Filaments, PHF) qui constituent les dégénérescences neurofibrillaires (DNF) de la maladie d'Alzheimer (Brion, 1985). Ceci a depuis été confirmé par de nombreuses équipes. Dans les années 1990, des modifications de Tau ont été rapportées dans plusieurs démences neurodégénératives qui sont maintenant regroupées sous le terme de « tauopathies » et dont la plus connue est la maladie d'Alzheimer (Buée et coll., 2002 ; Sergeant et coll., 2005).
). En réalité, elle favorise la polymérisation et la stabilité des microtubules. Depuis 1985, elle est aussi identifiée comme le composant majeur des paires de filaments en hélice (Paired Helical Filaments, PHF) qui constituent les dégénérescences neurofibrillaires (DNF) de la maladie d'Alzheimer (Brion, 1985). Ceci a depuis été confirmé par de nombreuses équipes. Dans les années 1990, des modifications de Tau ont été rapportées dans plusieurs démences neurodégénératives qui sont maintenant regroupées sous le terme de « tauopathies » et dont la plus connue est la maladie d'Alzheimer (Buée et coll., 2002 ; Sergeant et coll., 2005).
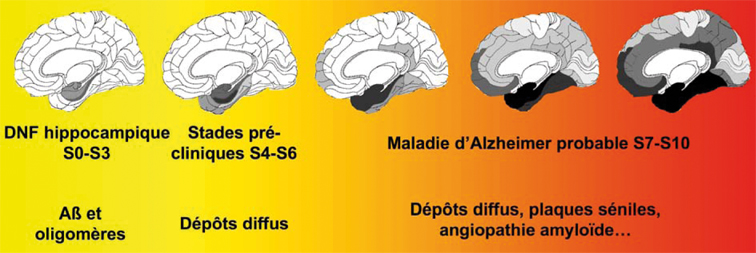
). En réalité, elle favorise la polymérisation et la stabilité des microtubules. Depuis 1985, elle est aussi identifiée comme le composant majeur des paires de filaments en hélice (Paired Helical Filaments, PHF) qui constituent les dégénérescences neurofibrillaires (DNF) de la maladie d'Alzheimer (Brion, 1985). Ceci a depuis été confirmé par de nombreuses équipes. Dans les années 1990, des modifications de Tau ont été rapportées dans plusieurs démences neurodégénératives qui sont maintenant regroupées sous le terme de « tauopathies » et dont la plus connue est la maladie d'Alzheimer (Buée et coll., 2002 ; Sergeant et coll., 2005). La maladie d'Alzheimer est caractérisée par la présence dans le cortex cérébral de deux lésions neuropathologiques particulières : les DNF et les dépôts de peptide Aβ (diffus ou amyloïdes) (figure 3.1). Les DNF ne sont pas spécifiques de la maladie d'Alzheimer. Des agrégats de protéines Tau sont retrouvés dans de nombreux syndromes parkinsoniens (dégénérescence cortico-basale, paralysie supranucléaire progressive, Parkinson postencéphalitique, syndrome de l'île de Guam), et certaines démences frontotemporales telles que la maladie de Pick, les démences frontotemporales associées à un syndrome parkinsonien liées au chromosome 17 (DFTP-17), une dystrophie myotonique comme la maladie de Steinert, et dans la région hippocampique au cours du vieillissement normal (pour revues, Buée et coll., 2000 ; Lee et coll., 2001 ; Sergeant et coll., 2005). Les affections dans lesquelles la protéine Tau s'accumule ont été regroupées sous le terme de « tauopathies ». Si les DNF ne sont pas caractéristiques de la maladie d'Alzheimer, leur association à une pathologie amyloïde et leur distribution topographique dans le cerveau sont spécifiques et reflètent un mécanisme particulier à la maladie d'Alzheimer. Ainsi, il existe des liens intimes entre la pathologie amyloïde et les neurones en DNF (Delacourte, 2006).
). Les DNF ne sont pas spécifiques de la maladie d'Alzheimer. Des agrégats de protéines Tau sont retrouvés dans de nombreux syndromes parkinsoniens (dégénérescence cortico-basale, paralysie supranucléaire progressive, Parkinson postencéphalitique, syndrome de l'île de Guam), et certaines démences frontotemporales telles que la maladie de Pick, les démences frontotemporales associées à un syndrome parkinsonien liées au chromosome 17 (DFTP-17), une dystrophie myotonique comme la maladie de Steinert, et dans la région hippocampique au cours du vieillissement normal (pour revues, Buée et coll., 2000 ; Lee et coll., 2001 ; Sergeant et coll., 2005). Les affections dans lesquelles la protéine Tau s'accumule ont été regroupées sous le terme de « tauopathies ». Si les DNF ne sont pas caractéristiques de la maladie d'Alzheimer, leur association à une pathologie amyloïde et leur distribution topographique dans le cerveau sont spécifiques et reflètent un mécanisme particulier à la maladie d'Alzheimer. Ainsi, il existe des liens intimes entre la pathologie amyloïde et les neurones en DNF (Delacourte, 2006). | Figure 3.1 Séquence d'apparition de la dégénérescence neurofibrillaire (DNF) et des dépôts de peptide Aβ au cours de la maladie d'Alzheimer |
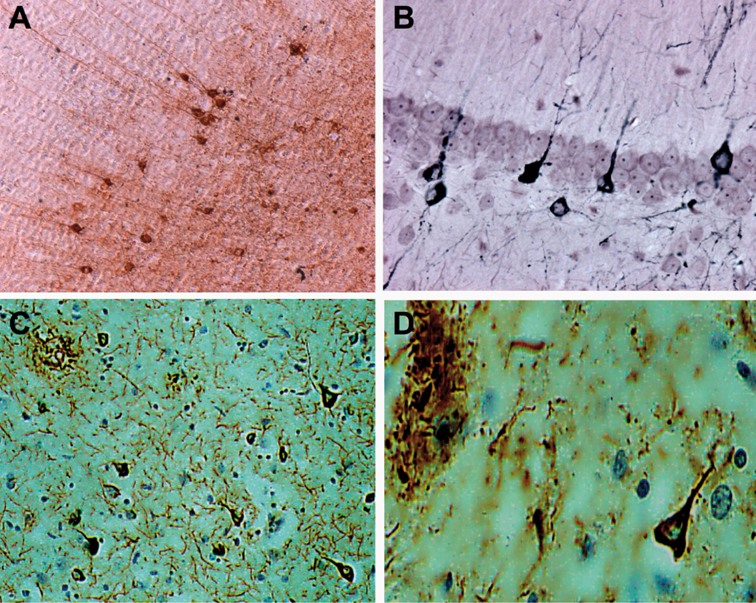
Les protéines Tau sont les constituants majeurs des filaments pathologiques intraneuronaux de la dégénérescence neurofibrillaire (Brion, 1985 ; Delacourte et Defossez, 1986) (figure 3.2). Les protéines Tau de la DNF sont agrégées et anormalement phosphorylées. Leur caractérisation biochimique par la technique des immuno-empreintes (« Western blot ») révèle la présence d'un triplet majeur de protéines phosphorylées (Tau 60, 64 et 69) accompagné d'un variant mineur à 72-74 kDa. L'ensemble de ces variants est également appelé A68 ou Tau-PHF (Paired Helical Filaments) (Delacourte et coll., 1990 ; Flament et Delacourte, 1990 ; Lee et coll., 1991).
; Delacourte et Defossez, 1986) (figure 3.2). Les protéines Tau de la DNF sont agrégées et anormalement phosphorylées. Leur caractérisation biochimique par la technique des immuno-empreintes (« Western blot ») révèle la présence d'un triplet majeur de protéines phosphorylées (Tau 60, 64 et 69) accompagné d'un variant mineur à 72-74 kDa. L'ensemble de ces variants est également appelé A68 ou Tau-PHF (Paired Helical Filaments) (Delacourte et coll., 1990 ; Flament et Delacourte, 1990 ; Lee et coll., 1991). | Figure 3.2 Immunomarquage obtenu avec un anticorps anti-Tau phosphorylé dans le cortex cérébral chez une souris transgénique Tau (A-B) et chez l'homme (C-D) |
Protéines Tau normales
Les protéines Tau appartiennent à la famille des MAP (Microtubule-Associated Proteins). Elles sont principalement neuronales et jouent un rôle dans la polymérisation des microtubules (pour revues, Cleveland, 1990 ; Delacourte et Buée, 1997).
; Delacourte et Buée, 1997).Le gène des protéines Tau est localisé sur le chromosome 17, à la position 17q21. Le transcrit primaire contient 16 exons (Andreadis et coll., 1992).Dans le cerveau, certains exons ne sont pas traduits. Les exons 2, 3 et 10 sont épissés de manière alternative et sont spécifiques du tissu cérébral adulte. L'épissage alternatif de ces 3 exons produit 6 combinaisons possibles (2-3-10- à 2+3+10+) (Goedert et coll., 1989a et b). Au niveau protéique, il y a donc six isoformes de protéines Tau dans le cerveau adulte. Il faut noter que l'expression des protéines Tau est régulée au cours du développement. Ainsi, une seule isoforme, dite fœ tale, est présente à la naissance et ne comporte pas d'inserts codés par les exons 2, 3 ou 10. Les autres isoformes apparaissent au cours du développement ultérieur. La longueur de leurs séquences varie de 352 à 441 acides aminés. Sur électrophorèse en gel de polyacrylamide en présence de sodium dodécyl sulfate (SDS-PAGE), les protéines Tau normales migrent entre 45 et 65 kDa (Goedert et coll., 1989a et b ; Himmler et coll., 1989). Il faut noter que, chez l'adulte, quatre isoformes de protéines Tau sont fortement exprimées et dites « majeures ». En revanche, les deux isoformes avec les séquences codées par les exons 2 et 3 sont présentes mais en plus faible quantité (ht410 et ht441).
Dans le cerveau, certains exons ne sont pas traduits. Les exons 2, 3 et 10 sont épissés de manière alternative et sont spécifiques du tissu cérébral adulte. L'épissage alternatif de ces 3 exons produit 6 combinaisons possibles (2-3-10- à 2+3+10+) (Goedert et coll., 1989a et b). Au niveau protéique, il y a donc six isoformes de protéines Tau dans le cerveau adulte. Il faut noter que l'expression des protéines Tau est régulée au cours du développement. Ainsi, une seule isoforme, dite fœ tale, est présente à la naissance et ne comporte pas d'inserts codés par les exons 2, 3 ou 10. Les autres isoformes apparaissent au cours du développement ultérieur. La longueur de leurs séquences varie de 352 à 441 acides aminés. Sur électrophorèse en gel de polyacrylamide en présence de sodium dodécyl sulfate (SDS-PAGE), les protéines Tau normales migrent entre 45 et 65 kDa (Goedert et coll., 1989a et b ; Himmler et coll., 1989). Il faut noter que, chez l'adulte, quatre isoformes de protéines Tau sont fortement exprimées et dites « majeures ». En revanche, les deux isoformes avec les séquences codées par les exons 2 et 3 sont présentes mais en plus faible quantité (ht410 et ht441).La partie amino-terminale des protéines Tau, encore appelée domaine de projection, a un rôle encore mal connu. Ce domaine de projection pourrait interagir avec la membrane plasmique et certains organites comme les mitochondries. Quant au domaine carboxy-terminal, comportant 3 (sans exon 10) ou 4 (avec exon 10) segments répétitifs, il contrôle la stabilité des microtubules. Les trois isoformes sans la séquence codée par l'exon 10 (10-) possèdent trois domaines de liaison aux microtubules (3R) et les 3 isoformes avec la séquence de l'exon 10 (10+) en ont quatre (4R). L'interaction avec les dimères de tubuline est plus forte avec ce quatrième domaine, ce qui stabilise davantage les microtubules et peut moduler la longueur des extensions neuritiques, ainsi que la plasticité neuronale (pour revue, Buée et coll., 2000).
).La phosphorylation est la principale modification post-traductionnelle des protéines Tau. Des 80 résidus sérine et thréonine de la protéine Tau, plus d'une trentaine se sont révélés être phosphorylés, en particulier, de part et d'autre des domaines de liaison aux microtubules. Il existe également cinq résidus tyrosine dont certains sont phosphorylés. La phosphorylation régule la stabilité des microtubules. Lorsqu'elle porte en particulier sur la région riche en prolines située en amont des motifs répétés, elle diminue l'affinité de la protéine Tau pour les microtubules, entraînant leur dépolymérisation. La phosphorylation des résidus sérine 262 et 356 (selon la numérotation de l'isoforme la plus longue), situés respectivement dans les premier et quatrième domaines de liaison, modulerait également l'affinité des protéines Tau aux microtubules.
Les kinases impliquées dans la phosphorylation de Tau sont nombreuses. Parmi les plus communes, citons les kinases dépendantes des cyclines (cdk), la GSK3β, les MAP kinases (Erk1/2, jun kinases JNKs et p38s), les MARK, la phosphorylase K, la pKA, la pKC et la Tau-tubuline kinase (Buée et coll., 2000 ; Avila, 2006). Il est aussi clairement établi qu'il y a une balance phosphorylation-déphosphorylation, la déphosphorylation dépendant des phosphatases 1, 2A, 2B et 5 (Iqbal et Grundke-Iqbal, 2005). La phosphorylation est également influencée par d'autres modifications post-traductionnelles comme la conformation de la liaison peptidique ou la glycosylation des résidus sérine ou thréonine. Ainsi, la peptidyl prolyl cis/trans isomérase Pin1 module la phosphorylation de Tau en facilitant son accessibilité à la phosphatase 2A (Zhou et coll., 2000 ; Galas et coll., 2006). La protéine Tau néosynthétisée peut être O-glycosylée par un groupe unique de N-acétyl glucosamine. Il existe un lien exclusif entre l'O-glycosylation et la phosphorylation. Ainsi, l'induction d'une hyperphosphorylation de Tau par inhibition de phosphatases s'associe à une diminution de l'O-glycosylation (Arnold et coll., 1996 ; Lefebvre et coll., 2003).
; Avila, 2006). Il est aussi clairement établi qu'il y a une balance phosphorylation-déphosphorylation, la déphosphorylation dépendant des phosphatases 1, 2A, 2B et 5 (Iqbal et Grundke-Iqbal, 2005). La phosphorylation est également influencée par d'autres modifications post-traductionnelles comme la conformation de la liaison peptidique ou la glycosylation des résidus sérine ou thréonine. Ainsi, la peptidyl prolyl cis/trans isomérase Pin1 module la phosphorylation de Tau en facilitant son accessibilité à la phosphatase 2A (Zhou et coll., 2000 ; Galas et coll., 2006). La protéine Tau néosynthétisée peut être O-glycosylée par un groupe unique de N-acétyl glucosamine. Il existe un lien exclusif entre l'O-glycosylation et la phosphorylation. Ainsi, l'induction d'une hyperphosphorylation de Tau par inhibition de phosphatases s'associe à une diminution de l'O-glycosylation (Arnold et coll., 1996 ; Lefebvre et coll., 2003).Protéines Tau pathologiques de la maladie d'Alzheimer
Différentes modifications post-traductionnelles peuvent affecter la protéine Tau chez les patients atteints de maladie d'Alzheimer.
Phosphorylation
Les protéines Tau sont les constituants principaux des filaments appariés en hélice (Paired Helical Filaments ou PHF) qui sont mis en évidence par l'examen en microscopie électronique des DNF. Des techniques d'isolement des PHF à partir d'homogénats de cortex cérébral provenant de patients atteints de maladie Alzheimer ont été mises au point. Les protéines Tau provenant de ces PHF sont anormalement phosphorylées. Après déphosphorylation, les Tau-PHF s'alignent avec les Tau normales, ce qui suggère que les 6 isoformes des protéines Tau sont phosphorylées. Une analyse plus fine des variants de protéines Tau de la maladie d'Alzheimer a conduit à identifier la correspondance entre isoformes et variants phosphorylés de Tau. L'immunoempreinte d'homogénats corticaux est caractérisée par un triplet majeur à 60 (Tau 60), 64 (Tau 64) et 69 (Tau 69) kDa et un variant mineur à 74 kDa (Tau 74) (Mulot et coll., 1994 ; Sergeant et coll., 1997a). Le variant mineur (Tau 74) correspond à l'hyperphosphorylation de l'isoforme la plus longue et la moins exprimée.
; Sergeant et coll., 1997a). Le variant mineur (Tau 74) correspond à l'hyperphosphorylation de l'isoforme la plus longue et la moins exprimée.On distingue la phosphorylation anormale et l'hyperphosphorylation de la protéine Tau (pour revues, Buée et coll., 2000 ; Lee et coll., 2001 ; Sergeant et coll., 2005). La phosphorylation anormale consiste en la phosphorylation en des sites qui dans des conditions physiologiques ne sont pas concernés par la phosphorylation. On parle d'épitope non-physiologique ; ce sont par exemple les épitopes reconnus par les anticorps AT100 et TG-3. En revanche, la protéine Tau est considérée comme hyperphosphorylée lorsqu'elle est phosphorylée au niveau d'épitopes physiologiques en plus grand nombre que dans un cerveau adulte normal ou lorsque pour un site donné un pourcentage élevé de protéine Tau est phosphorylé.
; Lee et coll., 2001 ; Sergeant et coll., 2005). La phosphorylation anormale consiste en la phosphorylation en des sites qui dans des conditions physiologiques ne sont pas concernés par la phosphorylation. On parle d'épitope non-physiologique ; ce sont par exemple les épitopes reconnus par les anticorps AT100 et TG-3. En revanche, la protéine Tau est considérée comme hyperphosphorylée lorsqu'elle est phosphorylée au niveau d'épitopes physiologiques en plus grand nombre que dans un cerveau adulte normal ou lorsque pour un site donné un pourcentage élevé de protéine Tau est phosphorylé. La phosphorylation anormale de Tau peut être visualisée par l'utilisation d'anticorps dirigés contre des sites de phosphorylation de Tau-PHF comme AT100 (pThr212/pSer214) (Hoffmann et coll., 1997 ; Mailliot et coll., 1998a ; Zheng-Fischhofer et coll., 1998), AP422 (pSer422) (Hasegawa et coll., 1996), 988 (pSer442) (Bussiere et coll., 1999), PHF-27 (pThr231/pSer235) (Hoffmann et coll., 1997), CP-3 (pSer214) et PG-5 (pSer409) (Jicha et coll., 1999) et TG-3 (pT231) (Jicha et coll., 1997b ; Hamdane et coll., 2003). D'autres anticorps dépendants de la phosphorylation reconnaissent une structure conformationnelle plus générale de la protéine Tau agrégée ; c'est le cas des anticorps MC1 (Jicha et coll., 1997b) et Alz-50 (Carmel et coll., 1996 ; Jicha et coll., 1997a).
; Mailliot et coll., 1998a ; Zheng-Fischhofer et coll., 1998), AP422 (pSer422) (Hasegawa et coll., 1996), 988 (pSer442) (Bussiere et coll., 1999), PHF-27 (pThr231/pSer235) (Hoffmann et coll., 1997), CP-3 (pSer214) et PG-5 (pSer409) (Jicha et coll., 1999) et TG-3 (pT231) (Jicha et coll., 1997b ; Hamdane et coll., 2003). D'autres anticorps dépendants de la phosphorylation reconnaissent une structure conformationnelle plus générale de la protéine Tau agrégée ; c'est le cas des anticorps MC1 (Jicha et coll., 1997b) et Alz-50 (Carmel et coll., 1996 ; Jicha et coll., 1997a). L'état de phosphorylation de Tau dépend de l'équilibre entre les kinases (qui phosphorylent) et les phosphatases (qui déphosphorylent).
Comme nous l'avons vu précédemment, de nombreuses kinases sont capables de phosphoryler la protéine Tau et d'interagir avec cette dernière. Parmi celles-ci, deux ont été purifiées avec la protéine Tau et ont été nommées protéines kinases de Tau (Tau protein kinases, TPK). On distingue la protéine kinase de Tau 1 (ou TPKI) qui fut ensuite identifiée comme la GSK3β (Glycogène Synthétase Kinase 3 Bêta) (Ishiguro et coll., 1992 et 1993). La seconde kinase nommée Tau protéine kinase 2 fut identifiée comme étant le complexe Cdk5/p25 (Takahashi et coll., 1991). Nous limiterons notre exposé à ces 2 kinases fortement impliquées dans la régulation physiologique et pathologique de Tau.
et 1993). La seconde kinase nommée Tau protéine kinase 2 fut identifiée comme étant le complexe Cdk5/p25 (Takahashi et coll., 1991). Nous limiterons notre exposé à ces 2 kinases fortement impliquées dans la régulation physiologique et pathologique de Tau.Protéine kinase de Tau 1 (TPKI)
Des fractions partiellement purifiées d'extraits cérébraux de patients atteints de maladie d'Alzheimer sont capables de provoquer une phosphorylation anormale de Tau. La TPKI fut isolée par co-purification avec les microtubules. La nature de cette interaction a récemment été élucidée, puisqu'il semble que la TPKI puisse lier la protéine Tau non phosphorylée sur son domaine de projection et qu'ainsi Tau lui serve de protéine d'ancrage sur les microtubules (Ishiguro et coll., 1992).
). Bien qu'il existe différents types de kinases capables de phosphoryler la protéine Tau, la protéine TPKI ou GSK3β semble jouer un rôle primordial dans la régulation de Tau (Ishiguro et coll., 1993). Ainsi, aussi bien dans des conditions physiologiques que pathologiques, TPKI est capable de phosphoryler Tau in vitro en 15 sites dont 2 de type non Ser/Thr-Pro. En outre, la TPKI peut phosphoryler Tau directement sur certains sites après une pré-phosphorylation de Tau par d'autres kinases. Par exemple, une pré-phosphorylation par TPKII serait capable d'augmenter de 9 fois la phosphorylation de la thréonine 231 par la TPKI. Ce site reconnu par l'anticorps AT180 influe sur la liaison de Tau aux microtubules. De façon inverse, la phosphorylation séquentielle de Tau par TPKI puis par la PKA va permettre l'apparition de l'épitope pathologique AT100 (Tau phosphorylée sur Thr212/Ser214) (Zheng-Fischhofer et coll., 1998). La genèse d'épitopes pathologiques de type PHF semble être un phénomène complexe puisque des kinases telles que la PKA, la DYRK et la PKN auront un effet opposé sur la capacité de TPKI à phosphoryler Tau. Elles pourront l'inhiber ou l'activer, selon les sites concernés et surtout selon l'ordre des événements de phosphorylation. La phosphorylation par la TPKI ne nécessite pas toujours l'intervention première d'une autre kinase. À titre d'exemple, la Ser-396 ou la Ser-404 peuvent être phosphorylées par la TPKI seule ; ce site est reconnu par PHF-1 (pour revues, Buée et coll., 2000 ; Avila et coll., 2004).
). Ainsi, aussi bien dans des conditions physiologiques que pathologiques, TPKI est capable de phosphoryler Tau in vitro en 15 sites dont 2 de type non Ser/Thr-Pro. En outre, la TPKI peut phosphoryler Tau directement sur certains sites après une pré-phosphorylation de Tau par d'autres kinases. Par exemple, une pré-phosphorylation par TPKII serait capable d'augmenter de 9 fois la phosphorylation de la thréonine 231 par la TPKI. Ce site reconnu par l'anticorps AT180 influe sur la liaison de Tau aux microtubules. De façon inverse, la phosphorylation séquentielle de Tau par TPKI puis par la PKA va permettre l'apparition de l'épitope pathologique AT100 (Tau phosphorylée sur Thr212/Ser214) (Zheng-Fischhofer et coll., 1998). La genèse d'épitopes pathologiques de type PHF semble être un phénomène complexe puisque des kinases telles que la PKA, la DYRK et la PKN auront un effet opposé sur la capacité de TPKI à phosphoryler Tau. Elles pourront l'inhiber ou l'activer, selon les sites concernés et surtout selon l'ordre des événements de phosphorylation. La phosphorylation par la TPKI ne nécessite pas toujours l'intervention première d'une autre kinase. À titre d'exemple, la Ser-396 ou la Ser-404 peuvent être phosphorylées par la TPKI seule ; ce site est reconnu par PHF-1 (pour revues, Buée et coll., 2000 ; Avila et coll., 2004). L'activation et l'accumulation de TPKI seraient des événements précoces de la DNF durant lesquels elle serait retrouvée co-localisée avec les protéines Tau hyperphosphorylées. Cette kinase serait alors majoritairement responsable de l'hyperphosphorylation de Tau au cours de la maladie d'Alzheimer (pour revues, Bhat et coll., 2004 ; Jope et Johnson, 2004).
; Jope et Johnson, 2004).La GSK3β semble aussi être impliquée dans la pathologie amyloïde de la maladie d'Alzheimer.
Protéine kinase de Tau 2 (TPKII)
La TPKII a été purifiée avec les microtubules issus de cerveaux bovins comme protéine phosphorylant Tau (Takahashi et coll., 1991) et son activité a été identifiée comme correspondant à celle de Cdk5 (Kobayashi et coll., 1993 ; Ishiguro et coll., 1994).
) et son activité a été identifiée comme correspondant à celle de Cdk5 (Kobayashi et coll., 1993 ; Ishiguro et coll., 1994).La protéine Cdk5 est l'homologue neuronal de la kinase Cdc2 ou Cdk1. Elle est d'ailleurs également nommée NCLK (neuronal Cdc2-like kinase) et c'est un membre de la famille des petites kinases Sérine/Thréonine dépendantes des cyclines.
La Cdk5 a été identifiée par son homologie avec la Cdc2 humaine (Lew et Wang, 1995). Elle possède 60 % d'identité avec la Cdc2. La Cdk5 est retrouvée dans tous les tissus, mais elle est fortement exprimée dans le système nerveux où son activité a été détectée (Tang et coll., 1996).
). Elle possède 60 % d'identité avec la Cdc2. La Cdk5 est retrouvée dans tous les tissus, mais elle est fortement exprimée dans le système nerveux où son activité a été détectée (Tang et coll., 1996).Elle est essentielle au développement du système nerveux central. Elle est impliquée dans le développement laminaire du cortex, dans la différenciation neuronale et dans la guidance axonale. De plus, Cdk5 est aussi impliquée dans la plasticité synaptique, la motilité et l'adhésion , ainsi que dans la neurodégénérescence (Kwon et Tsai, 2000 ; Tsai et coll., 2004).
; Tsai et coll., 2004).La TPKII et la Cdk5 ont été décrites comme étant associées aux PHF in vivo et comme étant capables de créer des épitopes des Tau-PHF. L'activité kinasique de Cdk5 est liée à l'expression spatiale, temporelle et à la localisation intracellulaire de p35. Ainsi, le clivage de p35 en p25 par la calpaïne s'associe à une dérégulation de l'activité de Cdk5. L'accumulation de p25 a été associée aux maladies neurodégénératives telles que la maladie d'Alzheimer et la SLA (sclérose latérale amyotrophique) (Tsai et coll., 2004).
).Le complexe Cdk5/p25 serait responsable de la phosphorylation mitotique rencontrée dans la maladie d'Alzheimer (Hamdane et coll., 2003) et pourrait expliquer la réactivation du cycle cellulaire dans la dégénérescence neuronale et l'apoptose (Hamdane et coll., 2005).
) et pourrait expliquer la réactivation du cycle cellulaire dans la dégénérescence neuronale et l'apoptose (Hamdane et coll., 2005).La kinase Cdk5 pourrait faire le lien entre les pathologies Tau et amyloïde (Lee et Tsai, 2003).
).Autres kinases
L'hyperphosphorylation des protéines Tau observée au cours de la maladie d'Alzheimer pourrait être liée soit à une augmentation de l'activité kinasique, soit à une diminution de l'activité des phosphatases (pour revue, Trojanowski et Lee, 1995). De nombreuses kinases ont été impliquées dans la phosphorylation pathologique de Tau (pour revue, Lovestone et Reynolds, 1997). Certaines kinases activées par le stress telles que les SAP kinases (Stress-Activated Protein kinases) telles que JNK/SAPK1, p38/SAPK2, SAPK3 ont été décrites comme phosphorylant Tau et semblent être de bons candidats pour la phosphorylation pathologique de cette protéine (Buée-Scherrer et Goedert, 2002). Parmi les différentes kinases actives dans la DNF, on retrouve des kinases mitotiques ou des kinases moins décrites telles que les CK1 (Casein Kinases) dont l'activité de l'isoforme delta est décrite comme pouvant augmenter jusqu'à 30 fois durant la maladie d'Alzheimer (Vincent et coll., 1997 ; Ghoshal et coll., 1999). Il faut noter que certaines protéines, par leur liaison à la protéine Tau, pourront favoriser son hyperphosphorylation ou au contraire protéger la protéine Tau de cette hyperphosphorylation. Le premier exemple est la protéine 14-3-3 zeta qui est retrouvée colocalisée avec la protéine Tau-PHF. La protéine 14-3-3 zeta serait capable par sa liaison avec la protéine Tau de promouvoir sa phosphorylation par la kinase PKA au niveau de la Ser262. La phosphorylation en ce site affecte fortement l'affinité de Tau pour les microtubules (Hashiguchi et coll., 2000). À l'inverse, certaines protéines telles que la protéine WOX1 (WW domain-containing oxidoreductase) pourront exercer un rôle dans la protection de Tau vis-à-vis de l'hyperphosphorylation. Ainsi, la déplétion de la protéine WOX1 in vivo aura pour conséquence la phosphorylation de Tau sur Thr231/Thr212 et les Ser515/Ser516. Elle induira aussi une stimulation de la phosphorylation de Tau par GSK3β ainsi que la formation d'agrégats de Tau. Enfin, l'expression de WOX1 est décrite comme inversement corrélée à la vulnérabilité des neurones à la DNF (Sze et coll., 2004).
). De nombreuses kinases ont été impliquées dans la phosphorylation pathologique de Tau (pour revue, Lovestone et Reynolds, 1997). Certaines kinases activées par le stress telles que les SAP kinases (Stress-Activated Protein kinases) telles que JNK/SAPK1, p38/SAPK2, SAPK3 ont été décrites comme phosphorylant Tau et semblent être de bons candidats pour la phosphorylation pathologique de cette protéine (Buée-Scherrer et Goedert, 2002). Parmi les différentes kinases actives dans la DNF, on retrouve des kinases mitotiques ou des kinases moins décrites telles que les CK1 (Casein Kinases) dont l'activité de l'isoforme delta est décrite comme pouvant augmenter jusqu'à 30 fois durant la maladie d'Alzheimer (Vincent et coll., 1997 ; Ghoshal et coll., 1999). Il faut noter que certaines protéines, par leur liaison à la protéine Tau, pourront favoriser son hyperphosphorylation ou au contraire protéger la protéine Tau de cette hyperphosphorylation. Le premier exemple est la protéine 14-3-3 zeta qui est retrouvée colocalisée avec la protéine Tau-PHF. La protéine 14-3-3 zeta serait capable par sa liaison avec la protéine Tau de promouvoir sa phosphorylation par la kinase PKA au niveau de la Ser262. La phosphorylation en ce site affecte fortement l'affinité de Tau pour les microtubules (Hashiguchi et coll., 2000). À l'inverse, certaines protéines telles que la protéine WOX1 (WW domain-containing oxidoreductase) pourront exercer un rôle dans la protection de Tau vis-à-vis de l'hyperphosphorylation. Ainsi, la déplétion de la protéine WOX1 in vivo aura pour conséquence la phosphorylation de Tau sur Thr231/Thr212 et les Ser515/Ser516. Elle induira aussi une stimulation de la phosphorylation de Tau par GSK3β ainsi que la formation d'agrégats de Tau. Enfin, l'expression de WOX1 est décrite comme inversement corrélée à la vulnérabilité des neurones à la DNF (Sze et coll., 2004). L'état de phosphorylation de la protéine Tau dépend de l'équilibre des activités kinases avec les activités phosphatases.
Phosphatases
Les protéines phosphatases 1, 2A, 2B et 5 ont été impliquées dans la régulation de la phosphorylation de Tau (Liu et coll., 2005b). L'activité des phosphatases serait diminuée dans la maladie d'Alzheimer (Gong et coll., 1993). De plus, l'ARNm de PP2A serait également sous-exprimé dans l'hippocampe de patients atteints de maladie d'Alzheimer (Vogelsberg-Ragaglia et coll., 2001).
). L'activité des phosphatases serait diminuée dans la maladie d'Alzheimer (Gong et coll., 1993). De plus, l'ARNm de PP2A serait également sous-exprimé dans l'hippocampe de patients atteints de maladie d'Alzheimer (Vogelsberg-Ragaglia et coll., 2001).L'inhibition des phosphatases pourrait être responsable de l'hyperphosphorylation et de la phosphorylation anormale de la protéine Tau. En effet, l'inhibition expérimentale des phosphatases par l'acide okadaïque provoque une phosphorylation anormale des protéines Tau ainsi que leur dissociation des microtubules associée à une apoptose neuronale (Harris et coll., 1993 ; Dupont-Wallois et coll., 1995 ; Arendt et coll., 1998 ; Mailliot et coll., 1998b ; Ksiezak-Reding et coll., 2000). De même, dans un modèle murin, l'hypothermie provoquée par l'hypoglycémie provoque une hyperphosphorylation de Tau en rapport avec une baisse de l'activité de la protéine phosphatase 2A relativement à Cdk5/p25 et GSK3β (Planel et coll., 2001 et 2004).
; Dupont-Wallois et coll., 1995 ; Arendt et coll., 1998 ; Mailliot et coll., 1998b ; Ksiezak-Reding et coll., 2000). De même, dans un modèle murin, l'hypothermie provoquée par l'hypoglycémie provoque une hyperphosphorylation de Tau en rapport avec une baisse de l'activité de la protéine phosphatase 2A relativement à Cdk5/p25 et GSK3β (Planel et coll., 2001 et 2004). En conséquence, un déséquilibre de la balance kinases/phosphatases semble suffire à une dérégulation de la protéine Tau du fait de son hyperphosphorylation et de sa phosphorylation anormale.
Isomérases
Les protéines phosphatases comme PP2A déphosphorylent leurs substrats lorsque la liaison peptidique suivant le résidu Ser/Thr phosphorylé est en conformation trans (Zhou et coll., 2000 ; Stukenberg et Kirschner, 2001). Certaines isomérases comme la peptidyl-prolyl isomérase Pin1 facilitent le passage de cis en trans et réciproquement (Lu, 2004 ; Landrieu et coll., 2006). Pin1 a un rôle important dans la différenciation neuronale (Hamdane et coll., 2006). Une dérégulation de l'activité de ces enzymes pourrait avoir des conséquences sur la phosphorylation de Tau (Galas et coll., 2006 ; Hamdane et coll., 2006) et son agrégation. En effet, les souris invalidées pour le gène de Pin1 développent avec l'âge des troubles moteurs et comportementaux associés à une hyperphosphorylation de la protéine Tau et à des DNF (Liou et coll., 2003). Ces résultats suggèrent que la protéine Pin1 aurait un rôle protecteur vis-à-vis de la DNF. Cette hypothèse est renforcée par le fait que l'expression de Pin1 dans les régions de l'hippocampe et du cortex pariétal est inversement corrélée à la vulnérabilité neuronale à la DNF (Liou et coll., 2003). Une accumulation cytoplasmique de Pin1 en granules a été décrite (Holzer et coll., 2002 ; Ramakrishnan et coll., 2003), mais celle-ci n'est co-localisée avec les protéines Tau que lors des stades précoces de la DNF. En effet, les formations granulaires de Pin1 sont co-localisées avec l'épitope précoce de phosphorylation anormale TG-3 (Ramakrishnan et coll., 2003). Cette accumulation granulaire de Pin1 semble spécifique de la pathologie Tau puisqu'elle est observée dans d'autres tauopathies telles que les mutations du gène de la protéine Tau, la maladie de Pick et plus rarement dans les cas de PSP (Ramakrishnan et coll., 2003).
; Stukenberg et Kirschner, 2001). Certaines isomérases comme la peptidyl-prolyl isomérase Pin1 facilitent le passage de cis en trans et réciproquement (Lu, 2004 ; Landrieu et coll., 2006). Pin1 a un rôle important dans la différenciation neuronale (Hamdane et coll., 2006). Une dérégulation de l'activité de ces enzymes pourrait avoir des conséquences sur la phosphorylation de Tau (Galas et coll., 2006 ; Hamdane et coll., 2006) et son agrégation. En effet, les souris invalidées pour le gène de Pin1 développent avec l'âge des troubles moteurs et comportementaux associés à une hyperphosphorylation de la protéine Tau et à des DNF (Liou et coll., 2003). Ces résultats suggèrent que la protéine Pin1 aurait un rôle protecteur vis-à-vis de la DNF. Cette hypothèse est renforcée par le fait que l'expression de Pin1 dans les régions de l'hippocampe et du cortex pariétal est inversement corrélée à la vulnérabilité neuronale à la DNF (Liou et coll., 2003). Une accumulation cytoplasmique de Pin1 en granules a été décrite (Holzer et coll., 2002 ; Ramakrishnan et coll., 2003), mais celle-ci n'est co-localisée avec les protéines Tau que lors des stades précoces de la DNF. En effet, les formations granulaires de Pin1 sont co-localisées avec l'épitope précoce de phosphorylation anormale TG-3 (Ramakrishnan et coll., 2003). Cette accumulation granulaire de Pin1 semble spécifique de la pathologie Tau puisqu'elle est observée dans d'autres tauopathies telles que les mutations du gène de la protéine Tau, la maladie de Pick et plus rarement dans les cas de PSP (Ramakrishnan et coll., 2003).En conséquence, l'isomérase Pin1 pourrait être un acteur précoce et important de la DNF aussi bien dans la maladie d'Alzheimer que dans d'autres tauopathies.
Outre la phosphorylation, de nombreuses modifications pathologiques ont été décrites pour la protéine Tau chez les patients atteints de la maladie d'Alzheimer. Parmi ces modifications post-traductionnelles, on retrouve l'ubiquitination, la glycation, l'oxydation ainsi que la protéolyse de Tau.
Ubiquitination
L'ubiquitine est une protéine de stress de 76 acides aminés. Elle est impliquée dans la dégradation dépendante de l'ATP des protéines à vie courte ou des protéines endommagées (pour revue, Petrucelli et Dawson, 2004). La présence de l'ubiquitine au niveau des inclusions de protéines Tau au cours de la maladie de Pick, de Parkinson ou de la maladie d'Alzheimer a été mise en évidence par des anticorps dirigés contre les formes conjuguées de l'ubiquitine (Mori et coll., 1987). L'ubiquitine est associée aussi bien aux plaques séniles qu'aux protéines Tau-PHFs. La densité d'ubiquitine associée aux lésions est corrélée avec la sévérité de la démence au cours de la maladie d'Alzheimer (He et coll., 1993). Une étude récente a montré que l'ubiquitine ligase CHIP était associée aux protéines Tau agrégées dans les PHFs et qu'elle était responsable de l'ubiquitination de Tau. La phosphorylation anormale de Tau précéderait son ubiquitination et le système chaperone Hsp70/CHIP serait responsable de la régulation de la demi-vie de Tau et de l'élimination sélective des protéines Tau anormales (Petrucelli et coll., 2004).
). La présence de l'ubiquitine au niveau des inclusions de protéines Tau au cours de la maladie de Pick, de Parkinson ou de la maladie d'Alzheimer a été mise en évidence par des anticorps dirigés contre les formes conjuguées de l'ubiquitine (Mori et coll., 1987). L'ubiquitine est associée aussi bien aux plaques séniles qu'aux protéines Tau-PHFs. La densité d'ubiquitine associée aux lésions est corrélée avec la sévérité de la démence au cours de la maladie d'Alzheimer (He et coll., 1993). Une étude récente a montré que l'ubiquitine ligase CHIP était associée aux protéines Tau agrégées dans les PHFs et qu'elle était responsable de l'ubiquitination de Tau. La phosphorylation anormale de Tau précéderait son ubiquitination et le système chaperone Hsp70/CHIP serait responsable de la régulation de la demi-vie de Tau et de l'élimination sélective des protéines Tau anormales (Petrucelli et coll., 2004).Glycation
La glycation est une modification post-traductionnelle impliquant une liaison covalente entre la partie aldéhydique d'un sucre et un groupe accepteur de type amino d'une protéine. Cette liaison covalente s'établit par une réaction non enzymatique de type Maillard qui conduit à la formation de produits hétérogènes finaux de glycation avancée (AGEs de la formule anglaise « Advanced Glycation End Products ») (Baynes et coll., 1989). Les altérations liées à la glycation peuvent avoir des conséquences aussi bien sur la structure de la protéine, sur sa fonction que sur sa dégradation. L'accumulation de produits hétérogènes finaux de glycation avancée (AGEs) a été observée au cours du vieillissement normal ainsi que dans de nombreuses maladies liées à l'âge comme la maladie d'Alzheimer (pour revue, Munch et coll., 1997). Ainsi, les plaques séniles aussi bien que les protéines Tau agrégées sont glyquées et cette glycation se fait essentiellement sur des résidus lysine(s) ou arginine(s). La présence des AGEs dans les PHFs suggère que la glycation a un rôle dans la physiopathologie de la dégénérescence neurofibrillaire (Yan et coll., 1995). De plus, les constituants des plaques séniles ainsi que des PHFs sont insolubles et résistent aux protéases : ces caractéristiques sont celles des AGEs. Enfin, la glycation de Tau peut s'effectuer au niveau de son domaine de liaison aux microtubules et elle potentialise ainsi l'agrégation de Tau (Nacharaju et coll., 1997 ; Ledesma et coll., 1998).
). Les altérations liées à la glycation peuvent avoir des conséquences aussi bien sur la structure de la protéine, sur sa fonction que sur sa dégradation. L'accumulation de produits hétérogènes finaux de glycation avancée (AGEs) a été observée au cours du vieillissement normal ainsi que dans de nombreuses maladies liées à l'âge comme la maladie d'Alzheimer (pour revue, Munch et coll., 1997). Ainsi, les plaques séniles aussi bien que les protéines Tau agrégées sont glyquées et cette glycation se fait essentiellement sur des résidus lysine(s) ou arginine(s). La présence des AGEs dans les PHFs suggère que la glycation a un rôle dans la physiopathologie de la dégénérescence neurofibrillaire (Yan et coll., 1995). De plus, les constituants des plaques séniles ainsi que des PHFs sont insolubles et résistent aux protéases : ces caractéristiques sont celles des AGEs. Enfin, la glycation de Tau peut s'effectuer au niveau de son domaine de liaison aux microtubules et elle potentialise ainsi l'agrégation de Tau (Nacharaju et coll., 1997 ; Ledesma et coll., 1998).Oxydation
L'oxydation semble participer à la fibrillogenèse de Tau. En effet, tout comme la phosphorylation, elle pourrait faciliter les premières étapes de l'agrégation. La dimérisation des protéines Tau pourrait s'effectuer par l'établissement de ponts disulfures intermoléculaires. Cette idée est confortée par le fait que la mutation de la cystéine 322 en alanine ou la présence d'un environnement réducteur inhibe l'agrégation in vitro (Schweers et coll., 1995 ; Friedhoff et coll., 1998). Cette Cys322 est située dans la partie C-terminale de la protéine Tau, région décrite comme essentielle à l'agrégation (Perez et coll., 1996). La présence d'une seconde cystéine en 291 dans les protéines Tau 4R permettrait la formation de ponts disulfures intramoléculaires et par conséquent l'apparition de dimères de Tau très stables. Cette caractéristique de la protéine Tau 4R la rendrait plus résistante à la fibrillogenèse. L'oxydation empêcherait la formation de ces ponts disulfures intramoléculaires confortant ainsi l'hypothèse de son implication dans l'agrégation de Tau. En conséquence, une oxydation associée à une hyperphosphorylation de la protéine Tau pourraient suffire par un effet synergique à la formation de filaments de protéine Tau (Perez et coll., 2000 ; Liu et coll., 2005a).
; Friedhoff et coll., 1998). Cette Cys322 est située dans la partie C-terminale de la protéine Tau, région décrite comme essentielle à l'agrégation (Perez et coll., 1996). La présence d'une seconde cystéine en 291 dans les protéines Tau 4R permettrait la formation de ponts disulfures intramoléculaires et par conséquent l'apparition de dimères de Tau très stables. Cette caractéristique de la protéine Tau 4R la rendrait plus résistante à la fibrillogenèse. L'oxydation empêcherait la formation de ces ponts disulfures intramoléculaires confortant ainsi l'hypothèse de son implication dans l'agrégation de Tau. En conséquence, une oxydation associée à une hyperphosphorylation de la protéine Tau pourraient suffire par un effet synergique à la formation de filaments de protéine Tau (Perez et coll., 2000 ; Liu et coll., 2005a).
Poly-glutamination
La TGase (Tissue transGlutaminase) est une enzyme dépendante du calcium. Elle catalyse la formation de liaisons covalentes entre des résidus de glutamine et une amine primaire d'une liaison peptidique liant des lysines ou des polyamines. Elle est normalement présente dans le neurone. Son activité ainsi que de son niveau d'expression sont augmentés au cours de la maladie d'Alzheimer (Johnson et coll., 1997). La TGase permettrait la liaison de plusieurs protéines entre elles, entraînant ainsi l'établissement de complexes protéiques insolubles et résistants à la dégradation (pour revues, Hoffner et Djian, 2005). Cette caractéristique a sous-tendu l'hypothèse selon laquelle la TGase pourrait être impliquée dans l'agrégation de la protéine Tau. Ainsi, in vitro la TGase permet la formation de filaments de Tau. Les protéines Tau agrégées issues de cerveaux de patients atteints de la maladie d'Alzheimer sont de plus immuno-réactives pour des anticorps dirigés contre la TGase. La poly-glutamination serait aussi impliquée dans l'agrégation de la protéine Tau dans d'autres tauopathies telles que la PSP (paralysie supra-nucléaire progressive) (Tucholski et coll., 1999 ; Zemaitaitis et coll., 2000 ; Singer et coll., 2002 ; Halverson et coll., 2005).
). La TGase permettrait la liaison de plusieurs protéines entre elles, entraînant ainsi l'établissement de complexes protéiques insolubles et résistants à la dégradation (pour revues, Hoffner et Djian, 2005). Cette caractéristique a sous-tendu l'hypothèse selon laquelle la TGase pourrait être impliquée dans l'agrégation de la protéine Tau. Ainsi, in vitro la TGase permet la formation de filaments de Tau. Les protéines Tau agrégées issues de cerveaux de patients atteints de la maladie d'Alzheimer sont de plus immuno-réactives pour des anticorps dirigés contre la TGase. La poly-glutamination serait aussi impliquée dans l'agrégation de la protéine Tau dans d'autres tauopathies telles que la PSP (paralysie supra-nucléaire progressive) (Tucholski et coll., 1999 ; Zemaitaitis et coll., 2000 ; Singer et coll., 2002 ; Halverson et coll., 2005).Protéolyse de Tau
L'hypothèse d'une protéolyse de Tau comme événement préalable à son agrégation est critiquable. En effet, la protéine Tau-PHF est plus résistante à la protéolyse que les protéines Tau normales (Wischik et coll., 1988 ; Yang et Ksiezak-Reding, 1995), adultes ou fœ tales rapidement protéolysées en particulier par la calpaïne. L'hyperphosphorylation des protéines Tau-PHFs est considérée comme un facteur majeur expliquant leur résistance à la protéolyse (Litersky et Johnson, 1992 ; Yang et Ksiezak-Reding, 1995 ; Yang et coll., 1997). Il faut noter enfin que ces calpaïnes sont activées par une augmentation du calcium intracellulaire et qu'au cours de la maladie d'Alzheimer, elles pourraient être aussi activées par le peptide amyloïde (Shea et coll., 1997).
; Yang et Ksiezak-Reding, 1995), adultes ou fœ tales rapidement protéolysées en particulier par la calpaïne. L'hyperphosphorylation des protéines Tau-PHFs est considérée comme un facteur majeur expliquant leur résistance à la protéolyse (Litersky et Johnson, 1992 ; Yang et Ksiezak-Reding, 1995 ; Yang et coll., 1997). Il faut noter enfin que ces calpaïnes sont activées par une augmentation du calcium intracellulaire et qu'au cours de la maladie d'Alzheimer, elles pourraient être aussi activées par le peptide amyloïde (Shea et coll., 1997).Cependant, de récentes études montrent qu'un clivage en C-terminal de la protéine Tau par la caspase-3 induit une modification structurale de la protéine qui serait reconnue par un anticorps MC1 (Rissman et coll., 2004). Cet anticorps est spécifique d'un épitope considéré comme un marqueur précoce de l'agrégation de Tau (Jicha et coll., 1997a). Le clivage de Tau par la caspase augmenterait sa capacité à s'agréger en modifiant sa conformation et en permettant l'interaction de ses extrémités N- et C-terminales (Rissman et coll., 2004). Ces données ont suggéré la cascade physiopathologique suivante : au cours de la maladie d'Alzheimer, le peptide Aβ ou un stress, par exemple oxydant, pourrait induire la protéolyse de Tau par activation des caspases et clivage au niveau Asp 421 (Gamblin et coll., 2003). La forme tronquée de Tau adopterait une conformation reconnue par l'anticorps MC1. L'acquisition de cette conformation induirait un début d'agrégation des protéines Tau complètes avec les formes tronquées, qui se produirait sur les microtubules – entraînant un blocage du transport microtubulaire. L'hyperphosphorylation des protéines Tau agrégées permettait de les détacher des microtubules et les protégerait du clivage par la caspase (Guillozet-Bongaarts et coll., 2006) mais aurait pour conséquence leur accumulation cytosolique, induisant à son tour une déstabilisation des microtubules (Rissman et coll., 2004). Il a, de plus, été rapporté que les produits de protéolyse de Tau favoriseraient l'apoptose (Chung et coll., 2001 ; Fasulo et coll., 2005) et participeraient à la fibrillogenèse de la protéine (Rohn et coll., 2002).
). Cet anticorps est spécifique d'un épitope considéré comme un marqueur précoce de l'agrégation de Tau (Jicha et coll., 1997a). Le clivage de Tau par la caspase augmenterait sa capacité à s'agréger en modifiant sa conformation et en permettant l'interaction de ses extrémités N- et C-terminales (Rissman et coll., 2004). Ces données ont suggéré la cascade physiopathologique suivante : au cours de la maladie d'Alzheimer, le peptide Aβ ou un stress, par exemple oxydant, pourrait induire la protéolyse de Tau par activation des caspases et clivage au niveau Asp 421 (Gamblin et coll., 2003). La forme tronquée de Tau adopterait une conformation reconnue par l'anticorps MC1. L'acquisition de cette conformation induirait un début d'agrégation des protéines Tau complètes avec les formes tronquées, qui se produirait sur les microtubules – entraînant un blocage du transport microtubulaire. L'hyperphosphorylation des protéines Tau agrégées permettait de les détacher des microtubules et les protégerait du clivage par la caspase (Guillozet-Bongaarts et coll., 2006) mais aurait pour conséquence leur accumulation cytosolique, induisant à son tour une déstabilisation des microtubules (Rissman et coll., 2004). Il a, de plus, été rapporté que les produits de protéolyse de Tau favoriseraient l'apoptose (Chung et coll., 2001 ; Fasulo et coll., 2005) et participeraient à la fibrillogenèse de la protéine (Rohn et coll., 2002).Agrégation de Tau et facteurs d'agrégation
Les caractéristiques de l'agrégation de la protéine Tau ont été essentiellement décrites par des études in vitro. La protéine Tau est flexible et soluble dans de nombreuses circonstances. Cependant, de nombreux fragments peptidiques de Tau s'agrègent facilement. Les séquences répétées semblent être impliquées dans la fibrillogenèse de Tau. Il faut noter que la séquence minimale de contact au cours de l'agrégation de Tau est comprise entre les acides aminés 317 et 335. En revanche, les acides aminés en N-terminal de la protéine inhiberaient la fibrillogenèse (pour revue, Buée et coll., 2000). La phosphorylation pourrait favoriser la dimérisation (Paudel, 1997).
). La phosphorylation pourrait favoriser la dimérisation (Paudel, 1997).Les polyanions peuvent favoriser l'agrégation des protéines Tau in vitro. Ces polyanions incluent l'héparine, d'autres types de GAG (glycosaminoglycanes), des ARN ou des acides polyglutamiques (Goedert et coll., 1996). Certains de ces polyanions, et notamment les ARN ou les GAG sulfatés, sont associés aux PHFs in vivo. Ils favorisent l'agrégation des Tau 3R et 4R et il semble que cette agrégation nécessite la présence d'au moins deux séquences répétées et de ponts disulfures (Friedhoff et coll., 2000).
). Certains de ces polyanions, et notamment les ARN ou les GAG sulfatés, sont associés aux PHFs in vivo. Ils favorisent l'agrégation des Tau 3R et 4R et il semble que cette agrégation nécessite la présence d'au moins deux séquences répétées et de ponts disulfures (Friedhoff et coll., 2000).Le processus de formation des PHFs est un processus lent nécessitant une forte concentration en protéines Tau.
Différents stades ont été décrits au cours de l'agrégation. Dans un premier temps, on observe une dimérisation des protéines par oxydation par des ponts disulfures (dimères stables), ou par des liaisons ioniques (dimères instables). La nucléation ultérieure, c'est-à-dire l'assemblage des dimères, est lente en l'absence de polyanions (ARN, poly-Glu, héparine) et fortement accélérée en leur présence. Enfin, il y a élongation qui dépend aussi des polyanions et de la stabilité des di-/oligo-mères (Sibille et coll., 2006).
).Il faut noter que l'agrégation de Tau est le fruit de plusieurs événements et que la participation des modifications post-traductionnelles pathologiques telles que l'oxydation, l'ubiquitination, la glycation, la glycosylation ou la protéolyse pourrait être nécessaire. De nombreux autres facteurs, comme l'ApoE, pourraient influencer la fibrillogenèse (Buée et coll., 2000).
). Protéines Tau des tauopathies
Dans des syndromes parkinsoniens comme le syndrome de l'île de Guam et le Parkinson post-encéphalitique, un triplet de protéines Tau similaire à celui de la maladie d'Alzheimer est observé (Hof et coll., 1994 ; Buée-Scherrer et coll., 1995 et 1997). Au contraire, la PSP a révélé un autre profil électrophorétique correspondant à la présence des variants Tau 64 et 69 et d'une forme mineure à 74 kDa. Ces variants sont également retrouvés dans la dégénérescence corticobasale. Ce profil reflète l'agrégation sélective d'isoformes de Tau à quatre domaines de liaison aux microtubules (Flament et coll., 1991 ; Buée-Scherrer et coll., 1996 ; Mailliot et coll., 1998b ; Sergeant et coll., 1999).
; Buée-Scherrer et coll., 1995 et 1997). Au contraire, la PSP a révélé un autre profil électrophorétique correspondant à la présence des variants Tau 64 et 69 et d'une forme mineure à 74 kDa. Ces variants sont également retrouvés dans la dégénérescence corticobasale. Ce profil reflète l'agrégation sélective d'isoformes de Tau à quatre domaines de liaison aux microtubules (Flament et coll., 1991 ; Buée-Scherrer et coll., 1996 ; Mailliot et coll., 1998b ; Sergeant et coll., 1999).Dans la maladie de Pick, deux autres variants appelés Tau 60 et 64, avec une forme mineure Tau 69, sont détectés. Les corps de Pick sont localisés principalement dans les couches II et VI de l'isocortex et les neurones granulaires du gyrus dentatus (Buée-Scherrer et coll., 1996). Ces neurones ne contiennent pas les isoformes de protéines Tau avec la séquence codée par l'exon 10 (Goedert et coll., 1989a). Or, seules les isoformes de protéines Tau hyperphosphorylées sans la séquence codée par l'exon 10 (trois domaines de liaison aux microtubules) présentent un tel profil électrophorétique. Il est donc clair que des isoformes sans la séquence codée par l'exon 10 s'agrègent au sein des corps de Pick ( Sergeant et coll., 1997b ; Delacourte et coll., 1998 ; Mailliot et coll., 1998b).
). Ces neurones ne contiennent pas les isoformes de protéines Tau avec la séquence codée par l'exon 10 (Goedert et coll., 1989a). Or, seules les isoformes de protéines Tau hyperphosphorylées sans la séquence codée par l'exon 10 (trois domaines de liaison aux microtubules) présentent un tel profil électrophorétique. Il est donc clair que des isoformes sans la séquence codée par l'exon 10 s'agrègent au sein des corps de Pick ( Sergeant et coll., 1997b ; Delacourte et coll., 1998 ; Mailliot et coll., 1998b). Il est possible de différencier certaines maladies neurodégénératives en fonction de leur profil électrophorétique Tau. Les différences biochimiques observées sont liées à la présence de différentes combinaisons d'isoformes de protéines Tau (trois ou quatre domaines de liaison aux microtubules). Le type d'isoforme, 3R ou 4R, trouvé dans les agrégats caractéristiques des diverses pathologies que nous avons considérées, pourrait dépendre du profil normal d'expression de ces isoformes dans les sous-populations sélectivement affectées. La notion de tauopathies, pathologies liées à l'agrégation spécifique d'isoformes de protéines, repose sur le fait que les isoformes de protéines Tau définissent des sous-populations neuronales spécifiques.
Génétique des tauopathies
La découverte de formes familiales de démences frontotemporales résultant de mutations sur le gène Tau permet de mieux comprendre comment les protéines Tau pourraient contribuer au dysfonctionnement et à la mort des neurones. Une quarantaine de mutations ont été identifiées chez des patients avec des symptômes cliniques et des caractéristiques neuropathologiques différents. Par ailleurs, un ensemble de mutations sur une région d'environ 1,3 Mb dans le gène de Tau a permis de définir deux haplotypes, nommés H1 et H2 (Gijselinck et coll., 2006). L'haplotype H1 a été associé à un risque élevé de développer une PSP (Rademakers et coll., 2005).
). L'haplotype H1 a été associé à un risque élevé de développer une PSP (Rademakers et coll., 2005).Lien direct entre une anomalie génétique de Tau et la pathologie : le cas des FTDP-17
Ces formes familiales autosomiques dominantes se caractérisent cliniquement par une démence de type frontal avec syndrome parkinsonien (FTDP17). D'un point de vue neuropathologique, elles sont généralement dépourvues de substance amyloïde. Cependant, de nombreux neurones comportant des DNF et des dépôts fibrillaires de type neuritique sont présents, avec comme constituants majeurs les protéines Tau hyperphosphorylées. Des mutations introniques et exoniques ont été localisées sur le gène de la protéine Tau à proximité ou au sein même des séquences codant pour les domaines de liaison aux microtubules dans différentes familles de FTDP-17. Ces résultats indiquent que les protéines Tau sont directement impliquées dans le processus pathologique conduisant à la mort cellulaire et aux signes cliniques (pour revues, Goedert et Jakes, 2005 ; Sergeant et coll., 2005 ; Pittman et coll., 2006).
; Sergeant et coll., 2005 ; Pittman et coll., 2006).
Les mutations trouvées sur le gène de Tau dans les régions introniques et parfois exoniques se trouvent à proximité des séquences codant les domaines de liaison aux microtubules (régions 3R ou 4R). La plupart de ces mutations conduisent à la surexpression des isoformes de Tau 4R et leur agrégation en filaments1
. L'équilibre entre les isoformes de Tau 3R et 4R est donc fondamental pour la physiologie neuronale. Ce changement dans la balance des isoformes de Tau conduit à des profils électrophorétiques particuliers en fonction des isoformes impliquées. Cependant, la pathologie est aussi modulée par de nombreux facteurs innés et acquis. C'est ainsi que pour une même mutation sur le gène de Tau, l'expression clinique dans une même famille peut être très différente (variation sur l'âge de début, durée de la maladie, profil clinique (Alzheimer, Pick, syndrome parkinsonien...) (van Swieten et coll., 2004).
).Lien indirect : épissage anormal dans la dystrophie myotonique de Steinert
La maladie de Steinert ou dystrophie myotonique de type 1 est une myopathie héréditaire. Elle est caractérisée par une dystrophie musculaire, une myotonie et la présence d'anomalies touchant de nombreux autres organes (yeux, cœ ur, atteinte respiratoire, gonades). La transmission est autosomique dominante (1 cas pour 7 500 naissances). L'anomalie génétique est liée à des répétitions excessives du codon CTG sur le bras long du chromosome 19, dans la partie 3' UTR de la DMPK (Dystrophy Myotonic Protein Kinase). Les répétitions de codons CTG sont transcrits mais non traduits. D'un point de vue neuropathologique, il existe des dégénérescences neurofibrillaires surtout nombreuses dans la formation hippocampique et le lobe temporal. Le profil électrophorétique des protéines Tau est particulier avec une bande majeure à 60 kDa (Vermersch et coll., 1996). L'épissage de l'exon 2 de Tau est altéré : les ARNm et les isoformes de Tau avec la séquence codée par cet exon sont fortement diminués (Sergeant et coll., 2001). Il en est de même pour l'exon 6 (Leroy et coll., 2006).
). L'épissage de l'exon 2 de Tau est altéré : les ARNm et les isoformes de Tau avec la séquence codée par cet exon sont fortement diminués (Sergeant et coll., 2001). Il en est de même pour l'exon 6 (Leroy et coll., 2006).Il est vraisemblable que des facteurs ou régulateurs d'épissage soient captés au sein des expansions de triplet CUG conduisant à une dérégulation des mécanismes d'épissage. Le neurone exprime donc un profil anormal d'isoformes de protéines Tau. Ici encore, ce déséquilibre dans la balance des isoformes est à l'origine des DNF. Les mêmes observations ont été rapportées récemment dans la dystrophie myotonique de type 2 (Maurage et coll., 2005).
).Synergie APP-Tau
De cette analyse, il apparaît que la pathologie Tau est liée de façon directe ou indirecte à la majorité des démences. Dans le cas de la maladie d'Alzheimer, il y a clairement un lien entre la pathologie Tau et le diagnostic clinique. Pourtant, des mutations dans les gènes codant pour les acteurs du métabolisme de la protéine précurseur du peptide amyloïde (APP) conduisent à la formation de peptide Aβ agrégé et à une toxicité neuronale. L'APP a donc un rôle déterminant et incontournable dans les formes familiales de la maladie d'Alzheimer, mais il existe encore de nombreuses zones d'ombre concernant les formes sporadiques. Il est clair que le dysfonctionnement de l'APP est au minimum un facteur de risque pour la maladie d'Alzheimer, voire la cause même de la cascade pathologique. Comme indiqué précédemment dans l'expertise, les dégénérescences neurofibrillaires sont présentes avant l'apparition des dépôts amyloïdes. L'analyse du tissu cérébral humain a permis de déterminer la séquence d'apparition des lésions (Braak et coll., 2006) et l'analyse biochimique, de comprendre que les anomalies biochimiques précèdent les lésions neuropathologiques. Ainsi, les DNF sont détectées de façon précoce, mais il existe déjà des anomalies dans le métabolisme de l'APP (Delacourte et coll., 2002) sans présence de dépôts amyloïdes. On peut donc envisager l'hypothèse suivante : la pathologie Tau est stimulée par le dysfonctionnement de l'APP conduisant à la propagation hiérarchisée de la dégénérescence neurofibrillaire à l'ensemble du cerveau. Cette hypothèse a pu être vérifiée dans un certain nombre de modèles animaux où la combinaison des pathologies Tau et amyloïde accélère le processus dégénératif (Gotz et coll., 2001 ; Lewis et coll., 2001). Cependant, la mécanistique de la synergie APP-Tau, mise en évidence par exemple dans la maladie de Parkinson, est encore mal comprise.
) et l'analyse biochimique, de comprendre que les anomalies biochimiques précèdent les lésions neuropathologiques. Ainsi, les DNF sont détectées de façon précoce, mais il existe déjà des anomalies dans le métabolisme de l'APP (Delacourte et coll., 2002) sans présence de dépôts amyloïdes. On peut donc envisager l'hypothèse suivante : la pathologie Tau est stimulée par le dysfonctionnement de l'APP conduisant à la propagation hiérarchisée de la dégénérescence neurofibrillaire à l'ensemble du cerveau. Cette hypothèse a pu être vérifiée dans un certain nombre de modèles animaux où la combinaison des pathologies Tau et amyloïde accélère le processus dégénératif (Gotz et coll., 2001 ; Lewis et coll., 2001). Cependant, la mécanistique de la synergie APP-Tau, mise en évidence par exemple dans la maladie de Parkinson, est encore mal comprise. Ceci a des conséquences évidentes pour les stratégies thérapeutiques dans la maladie d'Alzheimer. Une majorité des thérapies actuellement en essai clinique restent focalisées sur le dysfonctionnement de l'APP et la cascade amyloïde (anti-amyloïde comme Alzhemed™, immunothérapie et inhibiteurs de β- et ɣ-secrétases). Cette approche, qui a sa logique, ne s'attaque pas à la dégénérescence neurofibrillaire. À ce titre, il a été tout récemment rapporté que la réduction des protéines Tau endogènes dans un modèle de souris transgénique APP diminuait de façon significative des dysfonctionnements neuronaux liés à l'accumulation de peptide Aβ et améliorait les déficits comportementaux associés (Roberson et coll., 2007).
).En conclusion,
les protéines Tau peuvent être considérées à la fois comme acteurs et marqueurs d'une forme de neurodégénérescence caractérisée par la présence de DNF ou d'accumulations gliales de protéine Tau. Dans tous les cas, l'agrégation et la phosphorylation anormale des protéines Tau sont les seules caractéristiques communes au processus de dégénérescence neurofibrillaire.
Leur rôle dans les neuropathologies est certainement encore sous-estimé puisque des microdélétions sur la région chromosomique du gène Tau viennent d'être identifiées dans les syndromes avec retard mental et malformation congénitale (Shaw-Smith et coll., 2006).
).L'expression d'isoformes de protéines Tau et de certaines kinases permet aussi de définir un phénotype cellulaire des sous-populations neuronales vulnérables à la pathologie neurodégénérative. Par ailleurs, il existe une composante génétique qui modifie directement ou indirectement l'épissage alternatif de Tau et, par conséquent, la proportion relative d'isoformes de Tau exprimées.
Au total, les dérèglements de la protéine Tau, au niveau de son expression, de sa phosphorylation ou de son agrégation conduisent toujours à un dysfonctionnement neuronal qui s'amplifie comme une réaction en chaîne. Il en résulte une atteinte des fonctions cérébrales en lien avec les régions touchées. La protéine Tau est ou devrait être une cible thérapeutique majeure puisqu'elle concerne non seulement la maladie d'Alzheimer mais aussi la plupart des patients déments et des maladies neurodégénératives.
Bibliographie
[1] allen b, ingram e, takao m, smith mj, jakes r. Abundant Tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301s Tau protein.
J Neurosci. 2002;
22:9340- 9351
[2] andreadis a, brown wm, kosik ks. Structure and novel exons of the human Tau gene.
Biochemistry. 1992;
31:10626- 10633
[3] arendt t, holzer m, bruckner mk, janke c, gartner u. The use of okadaic acid in vivo and the induction of molecular changes typical for Alzheimer’s disease.
Neuroscience. 1998;
85:1337- 1340
[4] arnold cs, johnson gv, cole rn, dong dl, lee m, hart gw. The microtubule-associated protein Tau is extensively modified with O-linked N-acetylglucosamine.
J Biol Chem. 1996;
271:28741- 28744
[5] avila j. Tau phosphorylation and aggregation in Alzheimer’s disease pathology.
FEBS Lett. 2006;
580:2922- 2927
[6] avila j, lucas jj, perez m, hernandez f. Role of Tau protein in both physiological and pathological conditions.
Physiol Rev. 2004;
84:361- 384
[7] baynes jw, watkins ng, fisher ci, hull cj, patrick js. The amadori product on protein: structure and reactions.
Prog Clin Biol Res. 1989;
304:43- 67
[8] bhat rv, budd haeberlein sl, avila j. Glycogen synthase kinase 3: a drug target for Cns therapies.
J Neurochem. 2004;
89:1313- 1317
[9] braak h, alafuzoff i, arzberger t, kretzschmar h, del tredici k. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry.
Acta Neuropathol (Berl). 2006;
112:389- 404
[10] brion jp. Mise en évidence immunologique de la protéine Tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer.
Arch Biol (Brux). 1985;
95:229- 235
[11] buée l, bussiere t, buée-scherrer v, delacourte a, hof pr. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders.
Brain Res Brain Res Rev. 2000;
33:95- 130
[12] buée l, hamdane m, delobel p, sambo av, begard s. Tau story: from frontotemporal dementia to other tauopathies.
J Soc Biol. 2002;
196:103- 108
[13] buée-scherrer v, buée l, hof pr, leveugle b, gilles c. Neurofibrillary degeneration in amyotrophic lateral sclerosis/parkinsonism-dementia complex of guam. immunochemical characterization of Tau proteins.
Am J Pathol. 1995;
146:924- 932
[14] buée-scherrer v, hof pr, buée l, leveugle b, vermersch p. Hyperphosphorylated Tau proteins differentiate corticobasal degeneration and pick’s disease.
Acta Neuropathol (Berl). 1996;
91:351- 359
[15] buée-scherrer v, buée l, leveugle b, perl dp, vermersch p. Pathological Tau proteins in postencephalitic parkinsonism: Comparison with Alzheimer’s disease and other neurodegenerative disorders.
Ann Neurol. 1997;
42:356- 359
[16] buée-scherrer v, goedert m. Phosphorylation of microtubule-associated protein Tau by stress-activated protein kinases in intact cells.
FEBS Lett. 2002;
515:151- 154
[17] burger née buch k, padberg f, nolde t, teipel sj, stubner s. Cerebrospinal fluid Tau protein shows a better discrimination in young old (<70 years) than in old old patients with Alzheimer’s disease compared with controls.
Neurosci Lett. 1999;
277:21- 24
[18] bussiere t, hof pr, mailliot c, brown cd, caillet-boudin ml. Phosphorylated serine422 on Tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration.
Acta Neuropathol (Berl). 1999;
97:221- 230
[19] carmel g, mager em, binder li, kuret j. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology.
J Biol Chem. 1996;
271:32789- 32795
[20] chung cw, song yh, kim ik, yoon wj, ryu br. Proapoptotic effects of Tau cleavage product generated by caspase-3.
Neurobiol Dis. 2001;
8:162- 172
[21] cleveland dw. Microtubule mapping.
Cell. 1990;
60:701- 702
[22] cleveland dw, hwo sy, kirschner mw. Purification of Tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin.
J Mol Biol. 1977;
116:207- 225
[23] delacourte a. The natural and molecular history of Alzheimer’s disease.
J Alzheimers Dis. 2006;
9:187- 194
[24] delacourte a, defossez a. Alzheimer’s disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments.
J Neurol Sci. 1986;
76:173- 186
[25] delacourte a, buée l. Normal and pathological Tau proteins as factors for microtubule assembly.
Int Rev Cytol. 1997;
171:167- 224
[26] delacourte a, flament s, dibe em, hublau p, sablonniere b. Pathological proteins Tau 64 and 69 are specifically expressed in the somatodendritic domain of the degenerating cortical neurons during Alzheimer’s disease. Demonstration with a panel of antibodies against Tau proteins.
Acta Neuropathol (Berl). 1990;
80:111- 117
[27] delacourte a, sergeant n, wattez a, gauvreau d, robitaille y. Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation.
Ann Neurol. 1998;
43:193- 204
[28] delacourte a, sergeant n, champain d, wattez a, maurage ca. Nonoverlapping but Synergetic Tau and App pathologies in sporadic Alzheimer’s disease.
Neurology. 2002;
59:398- 407
[29] dupont-wallois l, sautiere pe, cocquerelle c, bailleul b, delacourte a, caillet-boudin ml. Shift from fetal-type to Alzheimer-type phosphorylated Tau proteins in Sknsh-Sy 5y cells treated with okadaic acid.
FEBS Lett. 1995;
357:197- 201
[30] fasulo l, ugolini g, cattaneo a. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by Tau Ftdp-mutation N279k.
J Alzheimers Dis. 2005;
7:3- 13
[31] flament s, delacourte a. Tau Marker?.
Nature. 1990;
346:22
[32] flament s, delacourte a, verny m, hauw jj, javoy-agid f. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type.
Acta Neuropathol (Berl). 1991;
81:591- 596
[33] friedhoff p, von bergen m, mandelkow em, davies p, mandelkow e. A nucleated assembly mechanism of alzheimer paired helical filaments.
Proc Natl Acad Sci USA. 1998;
95:15712- 15717
[34] friedhoff p, von bergen m, mandelkow em, mandelkow e. Structure of Tau protein and assembly into paired helical filaments.
Biochim Biophys Acta. 2000;
1502:122- 132
[35] galas mc, dourlen p, begard s, ando k, blum d. The peptidylprolyl Cis/Trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons: Implication in a pathological mechanism related to Alzheimer disease.
J Biol Chem. 2006;
281:19296- 19304
[36] gamblin tc, chen f, zambrano a, abraha a, lagalwar s. Caspase cleavage of Tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease.
Proc Natl Acad Sci USA. 2003;
100:10032- 10037
[37] ghoshal n, smiley jf, demaggio aj, hoekstra mf, cochran ej. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease.
Am J Pathol. 1999;
155:1163- 1172
[38] gijselinck i, bogaerts v, rademakers r, van der zee j, van broeckhoven c, cruts m. Visualization of mapt inversion on stretched chromosomes of Tau-negative frontotemporal dementia patients.
Hum Mutat. 2006;
27:1057- 1059
[39] goedert m, jakes r. Mutations causing neurodegenerative tauopathies.
Biochim Biophys Acta. 2005;
1739:240- 250
[40] goedert m, spillantini mg, jakes r, rutherford d, crowther ra. Multiple isoforms of human microtubule-associated protein Tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease.
Neuron. 1989a;
3:519- 526
[41] goedert m, spillantini mg, potier mc, ulrich j, crowther ra. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein Tau containing four tandem repeats: Differential expression of Tau protein Mrnas in human brain.
Embo J. 1989b;
8:393- 399
[42] goedert m, jakes r, spillantini mg, hasegawa m, smith mj, crowther ra. Assembly of microtubule-associated protein Tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans.
Nature. 1996;
383:550- 553
[43] gong cx, singh tj, grundke-iqbal i, iqbal k. Phosphoprotein phosphatase activities in Alzheimer disease brain.
J Neurochem. 1993;
61:921- 927
[44] gotz j, chen f, van dorpe j, nitsch rm. Formation of neurofibrillary tangles in P301l Tau transgenic mice induced by Abeta 42 fibrils.
Science. 2001a;
293:1491- 1495
[45] guillozet-bongaarts al, cahill me, cryns vl, reynolds mr, berry rw, binder li. Pseudophosphorylation of Tau at Serine 422 inhibits caspase cleavage: In vitro evidence and implications for tangle formation in vivo.
J Neurochem. 2006;
97:1005- 1014
[46] halverson ra, lewis j, frausto s, hutton m, muma na. Tau protein is cross-linked by transglutaminase in P301l Tau transgenic mice.
J Neurosci. 2005;
25:1226- 1233
[47] hamdane m, sambo av, delobel p, begard s, violleau a. Mitotic-like Tau phosphorylation by P25-Cdk5 kinase complex.
J Biol Chem. 2003;
278:34026- 34034
[48] hamdane m, bretteville a, sambo av, schindowski k, begard s. P25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death.
J Cell Sci. 2005;
118:1291- 1298
[49] hamdane m, dourlen p, bretteville a, sambo av, ferreira s. Pin1 allows for differential tau dephosphorylation in neuronal cells.
Mol Cell Neurosci. 2006;
32:155- 160
[50] harris ka, oyler ga, doolittle gm, vincent i, lehman ra. Okadaic acid induces hyperphosphorylated forms of Tau protein in human brain slices.
Ann Neurol. 1993;
33:77- 87
[51] hasegawa m, jakes r, crowther ra, lee vm, ihara y, goedert m. Characterization of Mab Ap422, a novel phosphorylation-dependent monoclonal antibody against Tau protein.
FEBS Lett. 1996;
384:25- 30
[52] hashiguchi m, sobue k, paudel hk. 14-3-3zeta is an effector of Tau protein phosphorylation.
J Biol Chem. 2000;
275:25247- 25254
[53] he y, duyckaerts c, delaere p, piette f, hauw jj. Alzheimer’s lesions labelled by anti-ubiquitin antibodies: Comparison with other staining techniques. A study of 15 cases with graded intellectual status in ageing and Alzheimer’s disease.
Neuropathol Appl Neurobiol. 1993;
19:364- 371
[54] himmler a, drechsel d, kirschner mw, martin dwjr. Tau Consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains.
Mol Cell Biol. 1989;
9:1381- 1388
[55] hof pr, nimchinsky ea, buée-scherrer v, buée l, nasrallah j. Amyotrophic Lateral Sclerosis/Parkinsonism-Dementia Complex of Guam: Quantitative neuropathology, immunohistochemical analysis of neuronal vulnerability, and comparison with related neurodegenerative disorders.
Acta Neuropathol (Berl). 1994;
88:397- 404
[56] hoffmann r, lee vm, leight s, varga i, otvos ljr. Unique Alzheimer’s disease paired helical filament specific epitopes involve double phosphorylation at specific sites.
Biochemistry. 1997;
36:8114- 8124
[57] hoffner g, djian p. Transglutaminase and diseases of the central nervous system.
Front Biosci. 2005;
10:3078- 3092
[58] holzer m, gartner u, stobe a, hartig w, gruschka h. inverse association of Pin1 and Tau accumulation in Alzheimer’s disease hippocampus.
Acta Neuropathol (Berl). 2002;
104:471- 481
[59] iqbal k, grundke-iqbal i. pharmacological approaches of neurofibrillary degeneration.
Curr Alzheimer Res. 2005;
2:335- 341
[60] ishiguro k, takamatsu m, tomizawa k, omori a, takahashi m. Tau protein kinase I converts normal Tau protein into A68-like component of paired helical filaments.
J Biol Chem. 1992;
267:10897- 10901
[61] ishiguro k, shiratsuchi a, sato s, omori a, arioka m. Glycogen synthase kinase 3 beta is identical to Tau protein kinase I generating several epitopes of paired helical filaments.
FEBS Lett. 1993;
325:167- 172
[62] ishiguro k, kobayashi s, omori a, takamatsu m, yonekura s. Identification of the 23 Kda subunit of Tau protein kinase II as a putative activator of Cdk5 in bovine brain.
FEBS Lett. 1994;
342:203- 208
[63] jicha ga, bowser r, kazam ig, davies p. Alz-50 and Mc-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant Tau.
J Neurosci Res. 1997a;
48:128- 132
[64] jicha ga, lane e, vincent i, otvos ljr, hoffmann r, davies p. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer’s disease.
J Neurochem. 1997b;
69:2087- 2095
[65] jicha ga, weaver c, lane e, vianna c, kress y. Camp-dependent protein kinase phosphorylations on Tau in Alzheimer’s disease.
J Neurosci. 1999;
19:7486- 7494
[66] johnson gv, cox tm, lockhart jp, zinnerman md, miller ml, powers re. Transglutaminase activity is increased in Alzheimer’s disease brain.
Brain Res. 1997;
751:323- 329
[67] jope rs, johnson gv. The glamour and gloom of glycogen synthase kinase-3.
Trends Biochem Sci. 2004;
29:95- 102
[68] kobayashi s, ishiguro k, omori a, takamatsu m, arioka m. A Cdc2-related kinase PSSALRE/Cdk5 is homologous with the 30 Kda subunit of Tau protein kinase II, a proline-directed Protein kinase associated with microtubule.
FEBS Lett. 1993;
335:171- 175
[69] ksiezak-reding h, he d, gordon-krajcer w, kress y, lee s, dickson dw. Induction of Alzheimer-specific Tau epitope At100 in apoptotic human fetal astrocytes.
Cell Motil Cytoskeleton. 2000;
47:236- 252
[70] kwon yt, tsai lh. The role of the P35/Cdk5 kinase in cortical development.
Results Probl Cell Differ. 2000;
30:241- 253
[71] landrieu i, smet c, wieruszeski jm, sambo av, wintjens r. Exploring the molecular function of Pin1 by nuclear magnetic resonance.
Curr Protein Pept Sci. 2006;
7:179- 194
[72] ledesma md, perez m, colaco c, avila j. Tau glycation is involved in aggregation of the protein but not in the formation of filaments.
Cell Mol Biol (Noisy-le-grand). 1998;
44:1111- 1116
[73] lee ms, tsai lh. Cdk5: One of the links between senile plaques and neurofibrillary tangles?.
J Alzheimers Dis. 2003;
5:127- 137
[74] lee vm, balin bj, otvos ljr, trojanowski jq. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau.
Science. 1991;
251:675- 678
[75] lee vm, goedert m, trojanowski jq. Neurodegenerative tauopathies.
Annu Rev Neurosci. 2001;
24:1121- 1159
[76] lefebvre t, caillet-boudin ml, buée l, delacourte a, michalski jc. O-Glcnac glycosylation and neurological disorders.
Adv Exp Med Biol. 2003;
535:189- 202
[77] leroy o, wang j, maurage ca, parent m, cooper t. Brain-specific change in alternative splicing of tau exon 6 in myotonic dystrophy type 1.
Biochim Biophys Acta. 2006;
1762:460- 467
[78] lew j, wang jh. Neuronal Cdc2-like kinase.
Trends Biochem Sci. 1995;
20:33- 37
[79] lewis j, dickson dw, lin wl, chisholm l, corral a. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant Tau and App.
Science. 2001;
293:1487- 1491
[80] liou yc, sun a, ryo a, zhou xz, yu zx. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration.
Nature. 2003;
424:556- 561
[81] litersky jm, johnson gv. Phosphorylation by camp-dependent protein kinase inhibits the degradation of Tau by calpain.
J Biol Chem. 1992;
267:1563- 1568
[82] liu q, smith ma, avila j, debernardis j, kansal m. Alzheimer-specific epitopes of Tau represent lipid peroxidation-induced conformations.
Free Radic Biol Med. 2005a;
38:746- 754
[83] liu f, grundke-iqbal i, iqbal k, gong cx. Contributions of protein phosphatases Pp1, Pp2a, Pp2b and Pp5 to the regulation of Tau phosphorylation.
Eur J Neurosci. 2005b;
22:1942- 1950
[84] lovestone s, reynolds ch. The phosphorylation of Tau: A critical stage in neurodevelopment and neurodegenerative processes.
Neuroscience. 1997;
78:309- 324
[85] lu kp. Pinning down cell signaling, cancer and Alzheimer’s disease.
Trends Biochem Sci. 2004;
29:200- 209
[86] lu kp, liou yc, vincent i. Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s disease.
Bioessays. 2003;
25:174- 181
[87] mailliot c, bussiere t, caillet-boudin ml, delacourte a, buée l. Alzheimer-specific epitope of At100 in transfected cell lines with Tau: toward an efficient cell model of tau abnormal phosphorylation.
Neurosci Lett. 1998a;
255:13- 16
[88] mailliot c, sergeant n, bussiere t, caillet-boudin ml, delacourte a, buée l. Phosphorylation of specific sets of Tau isoforms reflects different neurofibrillary degeneration processes.
FEBS Lett. 1998b;
433:201- 204
[89] maurage ca, udd b, ruchoux mm, vermersch p, kalimo h. Similar brain Tau pathology in Dm2/Promm and Dm1/Steinert disease.
Neurology. 2005;
65:1636- 1638
[90] mori h, kondo j, ihara y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease.
Science. 1987;
235:1641- 1644
[91] mulot sf, hughes k, woodgett jr, anderton bh, hanger dp. Phf-Tau from Alzheimer’s brain comprises four species on SDS-Page which can be mimicked by in vitro phosphorylation of human brain Tau by glycogen synthase kinase-3 beta.
FEBS Lett. 1994;
349:359- 364
[92] munch g, thome j, foley p, schinzel r, riederer p. Advanced glycation endproducts in ageing and Alzheimer’s disease.
Brain Res Brain Res Rev. 1997;
23:134- 143
[93] nacharaju p, ko l, yen sh. Characterization of in vitro glycation sites of Tau.
J Neurochem. 1997;
69:1709- 1719
[94] paudel hk. Phosphorylation by neuronal Cdc2-like protein kinase promotes dimerization of Tau protein in vitro.
J Biol Chem. 1997;
272:28328- 28334
[95] perez m, valpuesta jm, medina m, montejo de garcini e, avila j. Polymerization of Tau into filaments in the presence of heparin: The minimal sequence required for Tau-Tau interaction.
J Neurochem. 1996;
67:1183- 1190
[96] perez m, cuadros r, smith ma, perry g, avila j. Phosphorylated, but not native, Tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal.
FEBS Lett. 2000;
486:270- 274
[97] petrucelli l, dawson tm. Mechanism of neurodegenerative disease: Role of the ubiquitin proteasome system.
Ann Med. 2004;
36:315- 320
[98] petrucelli l, dickson d, kehoe k, taylor j, snyder h. Chip and Hsp70 regulate Tau ubiquitination, degradation and aggregation.
Hum Mol Genet. 2004;
13:703- 714
[99] pittman am, fung hc, de silva r. Untangling the Tau gene association with neurodegenerative disorders.
Hum Mol Genet. 2006;
15:R188- 195
[100] planel e, yasutake k, fujita sc, ishiguro k. Inhibition of protein phosphatase 2a overrides Tau protein kinase i/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in Tau hyperphosphorylation in the hippocampus of starved mouse.
J Biol Chem. 2001;
276:34298- 34306
[101] planel e, miyasaka t, launey t, chui dh, tanemura k. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease.
J Neurosci. 2004;
24:2401- 2411
[102] rademakers r, melquist s, cruts m, theuns j, del-favero j. High-density Snp haplotyping suggests altered regulation of Tau gene expression in progressive supranuclear palsy.
Hum Mol Genet. 2005;
14:3281- 3292
[103] ramakrishnan p, dickson dw, davies p. Pin1 colocalization with phosphorylated Tau in Alzheimer’s disease and other tauopathies.
Neurobiol Dis. 2003;
14:251- 264
[104] rissman ra, poon ww, blurton-jones m, oddo s, torp r. Caspase-cleavage of Tau is an early event in Alzheimer disease tangle pathology.
J Clin Invest. 2004;
114:121- 130
[105] roberson ed, scearce-levie k, palop jj, yan f, cheng ih. Reducing endogenous Tau ameliorates amyloid β–induced deficits in an Alzheimer’s disease mouse model.
Science. 2007;
316:DOI: 10.1126/science.1141736-
[106] rohn tt, rissman ra, davis mc, kim ye, cotman cw, head e. Caspase-9 activation and caspase cleavage of Tau in the Alzheimer’s disease brain.
Neurobiol Dis. 2002;
11:341- 354
[107] schweers o, mandelkow em, biernat j, mandelkow e. Oxidation of Cysteine-322 in the repeat domain of microtubule-associated protein Tau controls the in vitro assembly of paired helical filaments.
Proc Natl Acad Sci USA. 1995;
92:8463- 8467
[108] sergeant n, david jp, goedert m, jakes r, vermersch p. Two-dimensional characterization of paired helical filament-Tau from Alzheimer’s disease: Demonstration of an additional 74-kDa component and age-related biochemical modifications.
J Neurochem. 1997a;
69:834- 844
[109] sergeant n, david jp, lefranc d, vermersch p, wattez a, delacourte a. Different distribution of phosphorylated Tau protein isoforms in Alzheimer’s and Pick’s diseases.
FEBS Lett. 1997b;
412:578- 582
[110] sergeant n, wattez a, delacourte a. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: Tau pathologies with exclusively "exon 10" isoforms.
J Neurochem. 1999;
72:1243- 1249
[111] sergeant n, sablonniere b, schraen-maschke s, ghestem a, maurage ca. Dysregulation of human brain microtubule-associated Tau Mrna maturation in myotonic dystrophy type 1.
Hum Mol Genet. 2001;
10:2143- 2155
[112] sergeant n, delacourte a, buée l. Tau Protein as a differential biomarker of tauopathies.
Biochim Biophys Acta. 2005;
1739:179- 97
[113] shaw-smith c, pittman am, willatt l, martin h, rickman l. Microdeletion encompassing mapt at chromosome 17q21.3 is associated with developmental delay and learning disability.
Nat Genet. 2006;
38:1032- 1037
[114] shea tb, prabhakar s, ekinci fj. Beta-Amyloid and ionophore A23187 evoke Tau hyperphosphorylation by distinct intracellular pathways: Differential involvement of the calpain/protein kinase C system.
J Neurosci Res. 1997;
49:759- 768
[115] sibille n, sillen a, leroy a, wieruszeski jm, mulloy b. Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy.
Biochemistry. 2006;
45:12560- 12572
[116] singer sm, zainelli gm, norlund ma, lee jm, muma na. Transglutaminase bonds in neurofibrillary tangles and paired helical filament Tau early in Alzheimer’s disease.
Neurochem Int. 2002;
40:17- 30
[117] stukenberg pt, kirschner mw. Pin1 acts catalytically to promote a conformational change in Cdc25.
Mol Cell. 2001;
7:1071- 1083
[118] sze ci, su m, pugazhenthi s, jambal p, hsu lj. Down-regulation of Ww domain-containing oxidoreductase induces Tau phosphorylation in vitro. A Potential Role in Alzheimer’s Disease.
J Biol Chem. 2004;
279:30498- 30506
[119] takahashi m, tomizawa k, ishiguro k, sato k, omori a. A novel brain-specific 25 Kda protein (P25) is phosphorylated by a Ser/Thr-Pro kinase (Tpk Ii) from Tau protein kinase fractions.
FEBS Lett. 1991;
289:37- 43
[120] tang d, lee ky, qi z, matsuura i, wang jh. Neuronal Cdc2-Like kinase: From cell cycle to neuronal function.
Biochem Cell Biol. 1996;
74:419- 429
[121] trojanowski jq, lee vm. Phosphorylation of paired helical filament Tau in Alzheimer’s disease neurofibrillary lesions: Focusing on phosphatases.
Faseb J. 1995;
9:1570- 1576
[122] tsai lh, lee ms, cruz j. Cdk5, a therapeutic target for Alzheimer’s disease?.
Biochim Biophys Acta. 2004;
1697:137- 142
[123] tucholski j, kuret j, johnson gv. Tau is modified by tissue transglutaminase in situ: Possible functional and metabolic effects of polyamination.
J Neurochem. 1999;
73:1871- 1880
[124] van swieten jc, rosso sm, van herpen e, kamphorst w, ravid r, heutink p. Phenotypic variation in frontotemporal dementia and parkinsonism linked to chromosome 17.
Dement Geriatr Cogn Disord. 2004;
17:261- 264
[125] vermersch p, sergeant n, ruchoux mm, hofmann-radvanyi h, wattez a. Specific Tau variants in the brains of patients with myotonic dystrophy.
Neurology. 1996;
47:711- 717
[126] vincent i, jicha g, rosado m, dickson dw. Aberrant expression of mitotic Cdc2/Cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain.
J Neurosci. 1997;
17:3588- 3598
[127] vincent i, zheng jh, dickson dw, kress y, davies p. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease.
Neurobiol Aging. 1998;
19:287- 296
[128] vogelsberg-ragaglia v, schuck t, trojanowski jq, lee vm. Pp2a Mrna expression is quantitatively decreased in Alzheimer’s disease hippocampus.
Exp Neurol. 2001;
168:402- 412
[129] wischik cm, novak m, thogersen hc, edwards pc, runswick mj. Isolation of a fragment of Tau derived from the core of the paired helical filament of Alzheimer’s disease.
Proc Natl Acad Sci USA. 1988;
85:4506- 4510
[130] yan sd, yan sf, chen x, fu j, chen m. Non-enzymatically glycated Tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide.
Nat Med. 1995;
1:693- 699
[131] yang ls, ksiezak-reding h. Calpain-induced proteolysis of normal human Tau and Tau associated with paired helical filaments.
Eur J Biochem. 1995;
233:9- 17
[132] yang ls, gordon-krajcer w, ksiezak-reding h. Tau released from paired helical filaments with formic acid or guanidine is susceptible to calpain-mediated proteolysis.
J Neurochem. 1997;
69:1548- 1558
[133] zemaitaitis mo, lee jm, troncoso jc, muma na. Transglutaminase-induced cross-linking of Tau Proteins in progressive supranuclear palsy.
J Neuropathol Exp Neurol. 2000;
59:983- 989
[134] zheng-fischhofer q, biernat j, mandelkow em, illenberger s, godemann r, mandelkow e. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody At100 and requires a paired-helical-filament-like conformation.
Eur J Biochem. 1998;
252:542- 552
[135] zhou xz, kops o, werner a, lu pj, shen m. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25c and Tau proteins.
Mol Cell. 2000;
6:873- 883
→ Aller vers SYNTHESE