Enjeux sociétaux des tests génétiques
2008
| ANALYSE |
10-
Surveillance et régulation de l’usage des tests génétiques
La littérature consacrée à l’organisation de l’offre de tests ainsi qu’à la régulation des usages et de la mise à disposition des tests est très hétérogène. Elle comprend des bilans d’expérience sur tel ou tel type de test rédigés le plus souvent par des praticiens ou des professionnels impliqués dans la réalisation des analyses ou dans les consultations génétiques, des recommandations de bonnes pratiques émanant de sociétés savantes ou d’administrations de santé publique, les rapports produits par des comités spécialisés crées à l’initiative des états ou des institutions internationales, et enfin une importante littérature de sciences sociales consacrée soit à l’analyse de dispositifs particuliers, soit à la discussion générale des problèmes de régulation des outils et procédures de la biomédecine. Compte tenu de cette hétérogénéité ainsi que de la nature normative de toute discussion sur la façon dont nous devons juger de l’intérêt et de l’apport d’une innovation biomédicale, la présentation ci-dessous ne doit pas être considérée comme un « état des savoirs » présentant le consensus d’une communauté de spécialistes mais comme une introduction à un débat qui, pour pouvoir être mené de façon fructueuse, doit être documenté et informé. En conséquence, nous avons pour ce chapitre pris le parti d’insister sur les expériences de réflexion sur le statut des tests génétiques menés dans d’autres pays, en l’occurrence en Grande-Bretagne et aux États-Unis.
Régulation des pratiques biomédicales
Le concept de régulation a de multiples significations. Il est employé aussi bien par les biologistes et les médecins que les juristes ou les spécialistes en sciences sociales. Utilisé dans le contexte des pratiques médicales et de santé, il renvoie le plus souvent à l’expérience du médicament, c’est-à-dire à la question de l’encadrement et du contrôle par l’État et les puissances publiques de l’accès ainsi que des modalités de distribution des substances thérapeutiques dont l’usage par des non-spécialistes est considéré comme potentiellement dangereux (Chauveau, 1999 ; Bonah et Rasmussen, 2005). La régulation désigne alors un ensemble de dispositifs légaux et administratifs (monopole d’exercice, prescription, règles d’étiquetage, procédures d’homologation) permettant de définir qui fabrique, distribue et utilise les agents thérapeutiques.
; Bonah et Rasmussen, 2005). La régulation désigne alors un ensemble de dispositifs légaux et administratifs (monopole d’exercice, prescription, règles d’étiquetage, procédures d’homologation) permettant de définir qui fabrique, distribue et utilise les agents thérapeutiques.
; Bonah et Rasmussen, 2005). La régulation désigne alors un ensemble de dispositifs légaux et administratifs (monopole d’exercice, prescription, règles d’étiquetage, procédures d’homologation) permettant de définir qui fabrique, distribue et utilise les agents thérapeutiques.À partir de cette acception juridique et professionnelle, le concept de régulation a acquis une signification plus large (Baszanger et coll., 2000; Cambrosio et Keating, 2003; Daemmerich 2004). On peut le comprendre comme l’ensemble des dispositifs, formels et informels, qui participent à la sélection des formes d’intervention médicale qui sont, dans un contexte donné, considérées comme utiles et nécessaires. La régulation des pratiques de diagnostic et de soin inclut donc une palette d’outils allant des registres de preuve et hiérarchies épistémiques aux décrets et procédures administratives en passant par les recommandations et avis émanant de l’activité des professionnels de santé.
; Cambrosio et Keating, 2003; Daemmerich 2004). On peut le comprendre comme l’ensemble des dispositifs, formels et informels, qui participent à la sélection des formes d’intervention médicale qui sont, dans un contexte donné, considérées comme utiles et nécessaires. La régulation des pratiques de diagnostic et de soin inclut donc une palette d’outils allant des registres de preuve et hiérarchies épistémiques aux décrets et procédures administratives en passant par les recommandations et avis émanant de l’activité des professionnels de santé.Une seconde remarque préalable est qu’en parallèle à cet élargissement, la question des régulations est devenue une cible permanente du débat public. La multiplication des controverses sur les effets et conditions de mise sur le marché de tel ou tel agent thérapeutique en est un bon indice (Pignarre, 2003; Angell, 2004; Chauveau, 2004; Goozner, 2004; Urfalino, 2005). Cette visibilité publique tient bien évidemment au fait que l’État et les administrations publiques sont des acteurs de plus en plus directs de l’évaluation biomédicale, en particulier dans un contexte de limitation de l’augmentation des dépenses de santé. Elle tient aussi à l’implication croissante des patients et usagers, que ce soit à titre individuel ou par le biais de collectifs, dans les questions d’accès, de qualité des soins et de choix des priorités de recherches (Callon et Rabeharisoa, 1999; Barbot, 2002).
; Angell, 2004; Chauveau, 2004; Goozner, 2004; Urfalino, 2005). Cette visibilité publique tient bien évidemment au fait que l’État et les administrations publiques sont des acteurs de plus en plus directs de l’évaluation biomédicale, en particulier dans un contexte de limitation de l’augmentation des dépenses de santé. Elle tient aussi à l’implication croissante des patients et usagers, que ce soit à titre individuel ou par le biais de collectifs, dans les questions d’accès, de qualité des soins et de choix des priorités de recherches (Callon et Rabeharisoa, 1999; Barbot, 2002).Dans cette perspective, un thème important des sciences sociales de la santé est l’analyse de la multiplication des règles, dispositifs de normalisation et formes d’encadrement des pratiques biomédicales. Au-delà des enjeux économiques, deux tendances socio-historiques lourdes ont ainsi été mises en avant : d’abord la formation d’un complexe biomédical prenant place dans une configuration plus générale de techno-science ; ensuite la place croissante accordée à la notion de risque, aussi bien dans l’analyse des facteurs de survenue des pathologies que dans la définition des cibles de l’intervention médicale.
L’idée selon laquelle les liens entre science et technologie sont multiples, que les mêmes acteurs et les mêmes projets articulent souvent les deux dimensions et qu’il est par conséquent difficile de tracer une frontière nette entre recherche et développement, de présenter les trajectoires d’innovation diagnostique ou thérapeutique sous la forme d’une succession d’étapes linéaires a d’importantes conséquences. Sociologues et historiens insistent ainsi sur le fait que la biomédecine contemporaine n’est pas la juxtaposition d’une science biologique et d’un art thérapeutique. Il s’agit plutôt (et ceci est particulièrement vrai depuis la seconde moitié du moitié du XXe siècle) d’un système de relations complexes entre biologie, médecine et industrie, un ordre négocié liant des mondes sociaux hétérogènes, dans lesquels interagissent les chercheurs de laboratoire, les praticiens hospitaliers, les ingénieurs de l’industrie, les investisseurs de capital risque, les responsables de santé publique, les militants associatifs (Sinding, 1991; Löwy, 1996; Marks, 1997; Aronowitz, 1998; Cooter et Pickstone, 2000).
; Löwy, 1996; Marks, 1997; Aronowitz, 1998; Cooter et Pickstone, 2000).Au-delà de cette mise en exergue du continuum liant sciences, techniques et soin, l’émergence des biotechnologies génétiques est à l’origine d’un faisceau d’interrogations spécifiques portant sur les évolutions très récentes de la recherche biomédicale. En effet, si le phénomène « nouvelles biotechnologies » renvoie incontestablement à l’émergence de nouveaux objets de recherche (génomes, gènes de prédisposition, protéome…) ainsi que de nouvelles formes de modélisation et d’expérimentation (transgenèse, systèmes bio-informatiques), il correspond aussi à une phase de reconfiguration profonde des rapports entre biologie, marché et politique. Deux aspects de ces changements ont suscité de nombreuses analyses. Il s’agit tout d’abord de l’évolution des formes de la propriété intellectuelle avec une remontée de l’appropriation vers des objets de plus en plus fondamentaux (brevets sur les séquences de gènes) et la constitution, autour de la gestion du capital-risque, d’un nouveau marché des connaissances techno-scientifiques (Dasgupta et David, 1994; Eisenberg, 1997; Foray, 1999; Coriat, 2002; Mirowski, 2002). C’est ensuite le cas de la constitution de nouvelles arènes publiques où sont discutées les questions de priorité de recherche et d’usage des connaissances, notamment à l’initiative d’acteurs « profanes » qui, à l’image des associations Sida, ne sont pas des professionnels de la biologie ou de la médecine mais qui ont su, à travers leur engagement, faire émerger des formes originales d’expertise (Epstein, 1996; Dalgalarondo, 2003; Dodier, 2003).
; Eisenberg, 1997; Foray, 1999; Coriat, 2002; Mirowski, 2002). C’est ensuite le cas de la constitution de nouvelles arènes publiques où sont discutées les questions de priorité de recherche et d’usage des connaissances, notamment à l’initiative d’acteurs « profanes » qui, à l’image des associations Sida, ne sont pas des professionnels de la biologie ou de la médecine mais qui ont su, à travers leur engagement, faire émerger des formes originales d’expertise (Epstein, 1996; Dalgalarondo, 2003; Dodier, 2003).De nombreux travaux récents portent sur la modélisation de ces changements et ont pour ambitions de prendre en compte des échelles intermédiaires entre l’analyse très locale de processus d’invention et d’innovation et l’exploration à grande échelle des contraintes institutionnelles ou économiques (Gibbons et coll., 1994; Callon, 1995; Etzkowitz et Leydesdorff, 1997; Joly, 2001; Pestre, 2003). Pour un certain nombre d’auteurs, les transformations récentes sont si importantes qu’elles justifient une analyse en termes de basculement global du système scientifique et technique. Pour d’autres analystes, les changements sont plus contradictoires, les formes de production des savoirs existant à un moment donné sont hétérogènes, variant d’un domaine à l’autre, d’un lieu à l’autre. Tous s’accordent toutefois pour souligner les effets du rapprochement entre science et marchés.
; Callon, 1995; Etzkowitz et Leydesdorff, 1997; Joly, 2001; Pestre, 2003). Pour un certain nombre d’auteurs, les transformations récentes sont si importantes qu’elles justifient une analyse en termes de basculement global du système scientifique et technique. Pour d’autres analystes, les changements sont plus contradictoires, les formes de production des savoirs existant à un moment donné sont hétérogènes, variant d’un domaine à l’autre, d’un lieu à l’autre. Tous s’accordent toutefois pour souligner les effets du rapprochement entre science et marchés.Une seconde transformation importante pour comprendre le statut contemporain des tests génétiques est la généralisation de la catégorie de risque de maladie, à la fois comme catégorie analytique et comme cible d’intervention (Aronowitz, 1998; Berlivet, 2000). Avant les années 1960, le terme n’apparaît que rarement dans la littérature médicale. Il se généralise dans les années 1970 et les années 1980 dans le cadre de débats épidémiologiques qui amènent à parler de facteurs de risque (par exemple, dans le cas de l’étiologie des cancers), de risque thérapeutique (à propos des effets secondaires des médicaments) ou encore de risque iatrogène (à propos des infections contractées à l’hôpital). Il a pris, dans le contexte des analyses génétiques, une dimension plus interne qu’externe (le risque est « incorporé ») et plus individuelle que populationnelle (l’équation des facteurs de risque est une équation « personnelle »).
; Berlivet, 2000). Avant les années 1960, le terme n’apparaît que rarement dans la littérature médicale. Il se généralise dans les années 1970 et les années 1980 dans le cadre de débats épidémiologiques qui amènent à parler de facteurs de risque (par exemple, dans le cas de l’étiologie des cancers), de risque thérapeutique (à propos des effets secondaires des médicaments) ou encore de risque iatrogène (à propos des infections contractées à l’hôpital). Il a pris, dans le contexte des analyses génétiques, une dimension plus interne qu’externe (le risque est « incorporé ») et plus individuelle que populationnelle (l’équation des facteurs de risque est une équation « personnelle »).Il existe, en sciences sociales, un vaste corpus de réflexion sur le risque technique. Dans le cas des risques sanitaires, les questionnements ont initialement privilégié le problème de la perception des risques (Douglas, 1986), c’est-à-dire la recherche des facteurs culturels et sociaux explicatifs de l’opposition entre d’une part le risque de maladie des professionnels, probabiliste, quantifié, nourri par la description de processus bio-pathologiques, et d’autre part le risque de maladie des profanes, qualitatif, chargé de jugement de valeurs, lié aux expériences personnelles. On tend aujourd’hui à privilégier une approche en termes de construction collective dans laquelle le risque est objectivé par des dispositifs aux frontières de l’expérimental et du politique ; de sorte qu’il est doté de multiples significations, pour les profanes comme pour les experts (Joly, 2001; Gilbert, 2003).
), c’est-à-dire la recherche des facteurs culturels et sociaux explicatifs de l’opposition entre d’une part le risque de maladie des professionnels, probabiliste, quantifié, nourri par la description de processus bio-pathologiques, et d’autre part le risque de maladie des profanes, qualitatif, chargé de jugement de valeurs, lié aux expériences personnelles. On tend aujourd’hui à privilégier une approche en termes de construction collective dans laquelle le risque est objectivé par des dispositifs aux frontières de l’expérimental et du politique ; de sorte qu’il est doté de multiples significations, pour les profanes comme pour les experts (Joly, 2001; Gilbert, 2003).Quatre façons de réguler les pratiques biomédicales
Compte tenu de ces multiples niveaux de production et d’usage des entités de la biomédecine, on ne peut pas espérer caractériser un pattern unique de régulation des pratiques biomédicales qui, à un moment donné et en un lieu donné épuiserait les logiques d’action des différents acteurs et que l’on pourrait définir soit par l’identification de l’objet à réguler (médicaments, appareils, indications), soit par celle des institutions responsables (agences, sociétés savantes, organismes payeurs). Pour caractériser les régulations biomédicales, il importe de combiner un ensemble de critères qui concernent aussi bien les acteurs privilégiés, les objectifs de la régulation, les critères de preuve mis en jeu ou retenus, les outils ou dispositifs qui vont permettre l’encadrement et l’intervention.
En partant de la littérature consacrée aux trajectoires des agents thérapeutiques, on peut proposer une distinction entre quatre grandes formes de régulation (Daemmerich, 2004; Gaudillière, 2007). Les trois premières ont été utilisées pour analyser les transformations de la médecine au cours du dernier siècle. La dernière est le résultat des interrogations sur les recompositions des vingt dernières années (tableau 10.I).
; Gaudillière, 2007). Les trois premières ont été utilisées pour analyser les transformations de la médecine au cours du dernier siècle. La dernière est le résultat des interrogations sur les recompositions des vingt dernières années (tableau 10.I).Tableau 10.I Quatre formes de régulation biomédicale
|
Professionnelle
|
Étatique
|
Industrielle
|
Consumériste-civique
|
|
|---|---|---|---|---|
|
Enjeux, valeurs
|
Compétence, compliance
|
Accès, efficacité, santé publique
|
Productivité, qualité, rentabilité
|
Choix et autonomie des individus, qualité de vie
|
|
Acteurs pivots
|
Sociétés savantes, experts
|
Agences sanitaires, comités
|
Entreprises, groupes d’intérêts
|
Groupes d’usagers, victimes, associations de patients
|
|
Régime de preuve
|
Pharmacologie, bonnes indications et conditions d’usage
|
Statistique, essai clinique contrôlé
|
Contrôle de qualité Études de marché, Analyses coûts-bénéfices
|
Épidémiologie d’observation, Analyses bénéfice-risque
|
|
Outils pour l’intervention
|
Pharmacopée, vade-mecum thérapeutiques, recommandations, ordonnances
|
Procédures de mise sur le marché, avis publics, étiquetage obligatoire
|
Essais standards, publicité scientifique, notices et emballages
|
Dispositifs d’enregistrement des effets indésirables, jurisprudence, articles de presse
|
|
Arène privilégiée
|
Académique
|
Réglementaire
|
Économique
|
Médiatique, politique
|
Dans la régulation professionnelle, le praticien est la figure dominante, à la fois cible et initiateur des interventions. La régulation est nécessaire parce que les agents thérapeutiques sont des produits complexes, dangereux, pour lesquels il est indispensable de définir des indications et des doses précises. Le danger thérapeutique vient de l’absence de prise en compte des bonnes conditions de prescription par les « mauvais » professionnels. La régulation passe d’abord par l’écriture des recommandations d’indication et d’usage à l’initiative des sociétés savantes de médecins et de pharmaciens. Les principaux outils légaux sont les monopoles de prescription et de distribution établis par les statuts professionnels.
Au cours du XXe siècle, en réponse à l’industrialisation de la production des agents thérapeutiques, cette délégation du pouvoir régulateur a été associée à une intervention directe des administrations sanitaires et de l’État. L’apparition de régulations étatiques renvoie aux méfiances de nombreux praticiens à l’encontre des mécanismes du marché pharmaceutique. Dans cette perspective, les industriels, soucieux d’accroître leurs marges de profit cherchent à convaincre les médecins d’utiliser leurs produits, même lorsque l’on ne sait pas s’ils sont efficaces ou dangereux. Les prescripteurs sont d’autant plus réceptifs au discours des producteurs qu’ils partagent souvent avec eux une image miracle de l’action thérapeutique. L’enjeu est donc non seulement de contrôler les toxicités et effets secondaires mais aussi d’administrer le jugement d’utilité. Les agences du médicament définissent ainsi les indications d’usage à partir d’un cadre « épidémiologique » qui privilégie les dispositifs d’essais statistiquement contrôlés. Les outils légaux de l’intervention sont les dispositifs d’autorisation de la mise sur le marché et les notices à destination des utilisateurs.
Parallèlement à cet encadrement étatique, le changement des conditions de production a conduit à l’apparition d’une régulation industrielle. Pour l’entreprise productrice, la cible de la régulation n’est pas le « mauvais » médecin mais le praticien routinier trop lent à adopter les innovations. Une partie de la régulation fonctionne « en interne » avec le développement des standards de qualité et des procédures de contrôle destinées à éviter les plaintes et retours de marchandise. Mais l’essentiel concerne la construction des marchés avec la recherche marketing, les études coûts/bénéfices, la « publicité scientifique » auprès des prescripteurs. Les outils de régulation sont donc d’une part les procédures du contrôle de qualité et d’autre part les dispositifs de communication, en particulier le système des visiteurs médicaux.
À ces formes classiques de régulation biomédicale, s’ajoute une nouvelle façon de gérer les usages des agents thérapeutiques qui fait écho aux transformations évoquées plus haut et modifie les formes professionnelle, étatique et industrielle de la régulation biomédicale sans pour autant les faire disparaître. Cette régulation « consumériste-civique » met au centre la figure d’un usager informé capable de prendre des décisions complexes dans des situations où les actions médicales présentent à la fois des avantages et des inconvénients, où bénéfices et risques font l’objet de controverses. Les arènes privilégiées de cette régulation sont les arènes médiatiques et judiciaires avec l’association de patients ou de consommateurs comme acteur privilégié. Le cadre de jugement combine l’analyse bénéfices/risques et les enquêtes observationnelles permettant une surveillance des pratiques après mise sur le marché. Les outils d’intervention sont différenciés. Dans la variante « civique », il s’agit du procès exemplaire destiné à faire jurisprudence ou de la campagne de presse poussant les administrations sanitaires à intervenir. Dans la variante « consumériste », il s’agit de mettre à disposition des personnes une information plurielle, complète, permettant une prise de décision optimale en fonction des besoins de chacun.
Exemple de la régulation des tests BRCA
Une bonne manière d’illustrer l’importance de ces formes de régulation dans la gestion contemporaine des tests génétiques est de partir d’un cas concret. L’exemple que nous avons choisi est celui des débats suscités, en Europe et aux États-Unis, par le développement des services assurant la recherche des mutations prédisposant au cancer du sein et de l’ovaire. Deux raisons plaident pour cela. Premièrement, l’histoire des gènes BRCA a joué un rôle « fondateur » dans la réflexion sur les problèmes posés par les usages médicaux de la génomique dans la mesure où il s’agissait d’un des premiers exemples de diagnostic de modifications génétiques portant sur une pathologie majeure, jusqu’ici considérée comme marginalement héréditaire, intervenant longtemps après qu’une analyse moléculaire présymptomatique soit possible. En particulier, c’est autour du statut des gènes BRCA, de leur appropriation précoce par une entreprise privée de biotechnologie américaine, de l’organisation des services pratiquant la recherche de mutations qu’ont initialement été discutées des questions telles que celles de l’organisation de l’offre, de la brevetabilité, du lien entre diagnostic moléculaire et consultation génétique (Inserm et FNCLCC, 1998; Julian-Reynier et coll., 2005). Une seconde raison plaidant pour ce choix est l’existence d’une littérature de sciences sociales qui a notamment entrepris de comparer dans différents pays les modalités de réalisation de ces tests (Bourret, 2005; Gaudillière et Löwy, 2005; Parthasarathy, 2005).
; Julian-Reynier et coll., 2005). Une seconde raison plaidant pour ce choix est l’existence d’une littérature de sciences sociales qui a notamment entrepris de comparer dans différents pays les modalités de réalisation de ces tests (Bourret, 2005; Gaudillière et Löwy, 2005; Parthasarathy, 2005).États-Unis
Un premier élément tient aux effets de ce qu’on peut appeler le « modèle start-up » d’exploitation des connaissances sur les gènes BRCA. Sur la base de ses résultats dans le séquençage de BRCA1, la firme Myriad Genetics, fondée par des chercheurs issus de l’université de l’Utah à Salt Lake City, a en effet réussi dès le milieu des années 1990 à constituer un monopole reposant sur l’obtention d’un panel très complet de brevets « BRCA » et sur une division du travail originale entre la start-up et ses partenaires de l’industrie pharmaceutique. À la première sont allés les droits exclusifs sur le développement des tests dérivés de la connaissance des séquences BRCA, aux seconds les droits sur leurs utilisations thérapeutiques (Gaudillière et Cassier, 2001). Poursuivant cette logique, Myriad Genetics a décidé de valoriser elle-même les applications diagnostiques et a pour cela mis sur pied une plate-forme automatisée. Celle-ci a été intégrée à une filiale spécialisée dans la réalisation des diagnostics moléculaires qui opère la recherche des mutations BRCA par séquençage total du gène. Grâce à un accord commercial avec la firme concurrente Oncormed qui spécifiait les conditions de rachat des droits sur BRCA2, Myriad Genetics a obtenu les moyens d’un contrôle complet sur l’offre de tests. Ceci s’est traduit par le fait que, aux États-Unis, les quelques centres académiques qui avaient, à la fin des années 1980, maintenu une activité de recherche de mutations à la frontière entre routine et recherche, comme l’université de Pennsylvanie, ont dû cesser leur activité sous la menace de procès en contrefaçon.
). Poursuivant cette logique, Myriad Genetics a décidé de valoriser elle-même les applications diagnostiques et a pour cela mis sur pied une plate-forme automatisée. Celle-ci a été intégrée à une filiale spécialisée dans la réalisation des diagnostics moléculaires qui opère la recherche des mutations BRCA par séquençage total du gène. Grâce à un accord commercial avec la firme concurrente Oncormed qui spécifiait les conditions de rachat des droits sur BRCA2, Myriad Genetics a obtenu les moyens d’un contrôle complet sur l’offre de tests. Ceci s’est traduit par le fait que, aux États-Unis, les quelques centres académiques qui avaient, à la fin des années 1980, maintenu une activité de recherche de mutations à la frontière entre routine et recherche, comme l’université de Pennsylvanie, ont dû cesser leur activité sous la menace de procès en contrefaçon.Ce monopole n’est qu’un aspect du cadre marchand dans lequel est organisée la pratique des tests « BRCA » aux États-Unis. Une seconde caractéristique est la disjonction entre suivi médical et réalisation technique de la recherche de mutation. Le dispositif créé par Myriad Genetics repose en effet sur la généralisation du système présidant à la réalisation de la plupart des examens biologiques. Comme un dosage de glucose, l’analyse d’ADN est définie comme une activité technique dont le principal enjeu est la validité analytique. Celle-ci est perçue comme d’autant mieux garantie que les services qui en ont la charge sont plus spécialisés. Pour les dirigeants de la firme, l’existence ou non d’un conseil génétique, les possibilités de suivi clinique, ou les motifs justifiant la recherche génétique ne relèvent pas du marché biotechnologique mais d’une autre sphère, celle de l’encadrement de l’activité médicale. La réalisation d’une recherche de mutation à partir d’un prélèvement de sang n’est donc soumis qu’à une condition : la signature de la demande d’analyse par un médecin.
Une troisième caractéristique de ce modèle d’organisation est l’accès direct aux personnes. Myriad Genetics a régulièrement recours aux campagnes de promotion de ses services directement auprès des utilisatrices potentielles. Au départ très agressives, les initiatives en direction des femmes potentiellement à risque ont cédé la place à des dispositifs plus informatifs, en particulier sur Internet. Les « clientes » y trouvent non seulement des indications générales sur le cancer du sein, les gènes BRCA ou les interventions susceptibles de réduire le risque, mais aussi les moyens (tables, formulaires et adresses) pour évaluer leur propre statut et organiser la mise en œuvre du dépistage génétique. Il y a là plus qu’un service publicitaire. Pour les responsables de la firme, le droit à l’information génétique est un droit essentiel des individus. L’offre marchande se trouve ainsi relayée par la demande d’autonomie des personnes. Les biotechnologies génétiques y contribuent en apportant de nouveaux moyens pour connaître et préserver leur « capital santé ». Dans cette perspective, la connaissance des risques et leur bonne gestion ne sont pas que des questions de santé publique. Ils constituent des enjeux individuels d’autant plus pressants que la couverture médicale est, aux États-Unis, majoritairement basée sur des contrats individualisés (sauf pour les populations les plus pauvres qui bénéficient de la couverture fédérale).
Aux États-Unis, les discussions sur le statut et la réalisation des tests « BRCA » ont essentiellement porté sur les conditions de mise en œuvre des tests, sur leur utilité, sur les modalités de gestion du risque. La première phase de débat, à partir de 1996, a été dominée par les initiatives des sociétés savantes et des organisations professionnelles.
Les recommandations de bonne pratique qui ont sans doute eu le plus d’écho sont celles de l’American Society for Clinical Oncology (ASCO, 1996). Elles avaient deux objectifs : d’une part fixer le seuil de risque à partir duquel la recherche de mutation pouvait être utile ; d’autre part définir les conditions de cette exploration. Incertitudes sur le suivi et consentement éclairé occupaient une large place. Le principal bénéfice prévu du diagnostic était de réduire l’inquiétude des personnes pour lesquelles on ne trouverait pas de mutation. Pour les personnes dont il fallait considérer qu’elles étaient à fort risque, l’ASCO demandait aux cancérologues de discuter du choix entre surveillance renforcée par mammographie et chirurgie préventive (ASCO, 1996). Parallèlement, la société insistait sur la nécessité de donner une information complète par le biais des formulaires de consentement éclairé.
). Elles avaient deux objectifs : d’une part fixer le seuil de risque à partir duquel la recherche de mutation pouvait être utile ; d’autre part définir les conditions de cette exploration. Incertitudes sur le suivi et consentement éclairé occupaient une large place. Le principal bénéfice prévu du diagnostic était de réduire l’inquiétude des personnes pour lesquelles on ne trouverait pas de mutation. Pour les personnes dont il fallait considérer qu’elles étaient à fort risque, l’ASCO demandait aux cancérologues de discuter du choix entre surveillance renforcée par mammographie et chirurgie préventive (ASCO, 1996). Parallèlement, la société insistait sur la nécessité de donner une information complète par le biais des formulaires de consentement éclairé.Toutes les instances d’évaluation qui se saisirent de la question des tests « BRCA » durant cette période ne réagirent pas de la même manière. D’autres corpus de recommandations ont été élaborés, par exemple par un groupe de réflexion sur la génomique établi par l’Université de Stanford (SPGES, 1998). Celui-ci portait un regard beaucoup plus critique sur les modalités de commercialisation des tests. Rassemblant des généticiens moléculaires, des spécialistes de sciences sociales ainsi que des associations de femmes actives dans le domaine de la santé, le groupe de Stanford considérait qu’un accès rapide aux tests était une priorité. Il laissait l’évaluation des critères de prescription à l’appréciation des oncologues. En revanche, il considérait qu’une régulation publique était indispensable pour lutter contre d’éventuelles discriminations en matière d’embauche ou d’assurance. Une régulation du marché était aussi jugée nécessaire pour éviter que la généralisation des tests ne finisse par avoir des effets négatifs en termes de santé publique. Ce d’autant plus que seule la chirurgie prophylactique constitue une mesure de prévention reconnue comme efficace.
). Celui-ci portait un regard beaucoup plus critique sur les modalités de commercialisation des tests. Rassemblant des généticiens moléculaires, des spécialistes de sciences sociales ainsi que des associations de femmes actives dans le domaine de la santé, le groupe de Stanford considérait qu’un accès rapide aux tests était une priorité. Il laissait l’évaluation des critères de prescription à l’appréciation des oncologues. En revanche, il considérait qu’une régulation publique était indispensable pour lutter contre d’éventuelles discriminations en matière d’embauche ou d’assurance. Une régulation du marché était aussi jugée nécessaire pour éviter que la généralisation des tests ne finisse par avoir des effets négatifs en termes de santé publique. Ce d’autant plus que seule la chirurgie prophylactique constitue une mesure de prévention reconnue comme efficace.Grande-Bretagne
Le dispositif introduit par le National Health Service (NHS) en Grande-Bretagne est très différent. Il s’agit d’un encadrement professionnel et étatique. Le NHS a en effet organisé centralement l’offre de tests génétiques (Donai et Rob, 2001). Ceux-ci sont réalisés par dix-huit centres qui dépendent des autorités régionales du service de santé. À cela s’ajoutent deux centres de référence nationaux (Manchester et Salisbury) responsables des tests les moins demandés ainsi que des vérifications et du contrôle de qualité (NHS, 2003). Il s’agit d’un dispositif public centralisé qui autorise une planification faisant une large place à l’évaluation des coûts et à leur mise en regard de ce que les tests peuvent apporter en matière de santé publique. Dans ce cadre, les généralistes sont les principaux « gate keepers », rédigeant la plupart des prescriptions.
). Ceux-ci sont réalisés par dix-huit centres qui dépendent des autorités régionales du service de santé. À cela s’ajoutent deux centres de référence nationaux (Manchester et Salisbury) responsables des tests les moins demandés ainsi que des vérifications et du contrôle de qualité (NHS, 2003). Il s’agit d’un dispositif public centralisé qui autorise une planification faisant une large place à l’évaluation des coûts et à leur mise en regard de ce que les tests peuvent apporter en matière de santé publique. Dans ce cadre, les généralistes sont les principaux « gate keepers », rédigeant la plupart des prescriptions.La construction de l’offre de tests « BRCA » n’a pas eu pour point de départ une discussion préalable à l’attribution d’une autorisation de mise sur le marché, ou un débat sur les interventions cliniques afférentes à l’identification des risques génétiques mais une évaluation des ressources matérielles et humaines que le NHS pouvait raisonnablement investir dans la réalisation des tests. Les tests « BRCA » se trouvent ainsi placés à la croisée de deux réseaux. Chaque centre régional du cancer est associé à un centre de génétique médicale qui pratique toutes les analyses génétiques. Le premier souci de l’administration du NHS a été de ne pas engorger ces centres avec un flot de recherche de mutations BRCA. On a donc défini a priori un volume de tests raisonnable du point de vue des besoins médicaux et du point de vue des infrastructures nécessaires pour ensuite définir le système de sélection adapté ; dans ce cas avec une stricte grille de classement des personnes en individus « à risque » faible, moyen ou élevé en fonction de considérations épidémiologiques.
Par rapport à la configuration américaine, l’avantage de cette planification est d’offrir un couplage fort entre accès aux tests et prise en charge. Les femmes identifiées à haut risque se voient offrir conseil génétique, visites régulières et intensification de la surveillance radiographique. Le principal défaut du système tient au caractère imposé de cette limitation de l’offre. Sa mise en place n’a reposé sur aucun débat public, sur aucune implication des associations de malades. Elle a seulement fait appel à la négociation entre managers du système de santé et spécialistes de la génétique et/ou du cancer.
France et comparaison internationale
Le schéma français d’offre de test et de réglementation fait l’objet de plusieurs autres chapitres dans le cadre de cette expertise. Rappelons simplement les traits dominants de la configuration « BRCA ». Premièrement, du fait du rôle joué par les centres anticancéreux dans la mise en place des consultations, les tests « BRCA » sont réalisés dans un contexte clinique selon un modèle « intégré ». C’est-à-dire qu’une même structure réalise l’analyse moléculaire, les consultations de conseil génétique pré- et post-analyse, le suivi psychologique et la prise en charge thérapeutique (si la recherche de mutation est faite sur une patiente déjà diagnostiquée) ou préventive (s’il s’agit d’un diagnostic présymptomatique appelant une surveillance renforcée). Deuxièmement, ce cadre clinique d’organisation de l’offre est allé de pair avec une relativement faible standardisation des pratiques. Chaque centre a développé ses propres compétences, procédures et innovations. Dans un premier temps, l’homogénéisation relative des protocoles et des choix de prise en charge a été le fait du groupe national d’onco-génétique, selon un modèle de régulation interne à la spécialité. Dans un second temps, l’administration de la santé a mis en place un système d’accréditation destiné à garantir les compétences techniques des centres réalisant des tests. Ainsi, c’est au niveau de la qualité des analyses moléculaires que porte la régulation étatique. Troisièmement, il n’existe pas de marché du test puisque ceux-ci sont intégrés à la pratique hospitalière. Initialement, ils ont été pris en charge dans le cadre du financement des protocoles de recherche. Depuis, le Plan cancer a rendu la prise en charge pérenne avec une ligne budgétaire spécifique. En conséquence, les conflits sur la propriété intellectuelle et le monopole revendiqué par Myriad Genetics ont, en France, été particulièrement vifs. Les cliniciens français considérant que le modèle « start-up » était une menace directe sur la poursuite de leur activité. La volonté de Myriad Genetics d’étendre son monopole de propriété intellectuelle à l’Europe a ainsi conduit l’Institut Curie, l’Assistance Publique et les centres anti-cancéreux à mettre en œuvre des procédures d’opposition devant l’European Patent Office (EPO) encore en cours, laquelle a déjà eu pour résultat l’annulation d’une partie des brevets de Myriad Genetics. Cette comparaison de la pratique des tests « BRCA » est résumée dans le tableau 10.II
Tableau 10.II Comparaison de la pratique des tests « BRCA »
|
États-Unis
|
Royaume-Uni
|
France
|
|
|---|---|---|---|
|
Qui fait ?
|
Opérateurs commerciaux, marché du test, accès « direct » des patientes
|
Opérateurs publics de test : centres régionaux de génétique, dépendants du National Health Service
|
Services intégrés d’oncogénétique dans les centres de lutte contre le cancer et certains CHU
|
|
Sur qui ?
|
Population large, définie par les cliniques du risque
|
Population restreinte définie par les optimisations coûts/bénéfices
|
Population à risque « héréditaire » fort
|
|
Pourquoi faire ?
|
Chirurgie préventive, surveillance renforcée (imagerie)
|
Surveillance renforcée (general practice + imagerie)
|
Surveillance renforcée (imagerie), chirurgie expérimentale
|
|
Qui paye ?
|
HMO*, assurances individuelles
|
National Health Service
|
Budgets recherche et plan « Génétique et cancer »
|
|
Quelles controverses ?
|
Discrimination dans l’accès ; utilité clinique
|
Tardives, sur l’accès
|
Propriété intellectuelle et monopole commercial
|
* Health Maintenance Organization
Si dans ces trois pays, il existe des éléments renvoyant à chacune des quatre formes de régulation évoquées plus haut, celles-ci n’y ont pas la même importance et ne jouent pas le même rôle. Au risque de la simplification, on peut considérer qu’aux États-Unis domine une combinaison de régulation industrielle et consumériste, en Grande-Bretagne une régulation professionnelle et étatique, en France une régulation professionnelle.
Évaluation d’utilité préalable à la mise sur le marché
Aux États-Unis, le débat, si ce n’est la pratique de la régulation des tests génétiques, ne se limite toutefois pas aux initiatives des associations médicales ou académiques. L’idée d’une régulation renforcée a principalement été défendue par les comités que l’administration fédérale de la santé a mis en place pour suivre l’expansion des tests génétiques.
La première initiative en ce sens a été la Task Force on Genetic Testing créée dans le cadre du programme génome des National Institutes of Health (NIH). En 1999, ce panel estimait nécessaire d’organiser un jugement collectif des bénéfices et risques en préalable à l’entrée en clinique courante des nouveaux tests (Holtzman et Watson, 1998). Cependant, cette Task Force s’avéra incapable de dégager un consensus sur les conditions de cette évaluation. Le Secretary’s Advisory Committee on Genetic Testing a été chargé en 1998 de la préparation d’une réglementation (SACGT, 1998).
). Cependant, cette Task Force s’avéra incapable de dégager un consensus sur les conditions de cette évaluation. Le Secretary’s Advisory Committee on Genetic Testing a été chargé en 1998 de la préparation d’une réglementation (SACGT, 1998).De composition hybride, incluant les institutions de santé publique (Food and Drug Administration ou FDA, Center for Diseases Control ou CDC), une grande variété d’organisations professionnelles (médicales ou biotechnologiques) et une représentation associative, ce comité a pendant deux ans organisé les consultations et le débat public sur la possibilité d’une régulation des tests génétiques par la FDA (Gaudillière et Cassier, 2001). À sa manière, le SACGT a constitué un nouveau lieu de « démocratie technique » (Callon et coll., 2001).
). À sa manière, le SACGT a constitué un nouveau lieu de « démocratie technique » (Callon et coll., 2001).En son sein, les développeurs de tests, biologistes moléculaires et entreprises de biotechnologies, se sont opposés à la perspective de régulation par une agence à partir de trois arguments : le droit à l’information des personnes, l’absence de spécificité des tests génétiques, le caractère satisfaisant des procédures existantes pour encadrer l’activité technique des laboratoires (les normes CLIA, proches du système français d’accréditation). Ainsi, la firme de biotechnologie Affymetrix déclara considérer « que le droit de l’individu à la connaissance de sa propre information génétique doit être protégé. (…) Nous avons les plus grandes réserves à l’endroit de toute recommandation introduisant un traitement particulier de l’information génétique. Nous pensons que les mêmes règles de confidentialité et de respect de la vie privée doivent régir toutes les informations médicales. Il n’existe pas de ligne de démarcation nette entre tests génétiques et autres sources courantes d’information médicale. Tenter d’établir une telle ligne de partage serait un véritable défi qui pourrait s’avérer contre-productif. Nous pensons qu’un traitement particulier de l’information génétique ne fera que renforcer les craintes du public à propos des abus et atteintes possibles à la vie privée. » (SACGT, 2000).
).Les associations de praticiens étaient beaucoup plus divisées. D’un côté, on trouvait l’association des conseillers génétiques (National Society of Genetic Counselors) très favorable à une régulation. De l’autre côté, l’American College of Medical Genetics rejoignait la position des biotechnologues en faveur d’un renforcement des normes CLIA. Entre les deux, l’American Society of Human Genetics souhaitait exempter de contrôle les tests de mutations pour des maladies rares, pratiqués par des institutions académiques.
Les associations de patients étaient toutes favorables à une régulation fédérale mais avec des motivations très hétérogènes. La plupart d’entre elles privilégiaient le contrôle de qualité et la garantie de non-discrimination en matière d’accès aux soins et d’assurance. Seule la coordination nationale des organisations de femmes pour la lutte contre le cancer du sein, la National Breast Cancer Coalition (NBCC) insistait sur la nécessité d’une évaluation spécifique de l’utilité clinique par une autorité indépendante (Casamayou, 2001). Pour cette organisation, celle-ci est d’autant plus nécessaire que l’exemple des tests « BRCA » montre comment un service commercial de test a été mis en œuvre sans évaluation préalable de son intérêt alors même que ni la surveillance radiologique, ni la chirurgie prophylactique, ni la prévention hormonale ne constituent des formes d’intervention satisfaisante. Dans la dernière en date de ses prises de position, le groupe « génétique » de la NBCC concluait ainsi : « Un test génétique pour le cancer du sein ne doit pas être utilisé sans une prise en compte complète de ses limites et de ses implications. Les tests apportent peu de bénéfices. (…) S’ils peuvent aider les femmes ayant une importante histoire familiale de cancer du sein et qui sont porteuses de mutations des gènes BRCA 1/2 à prendre des décisions, il faut se souvenir que les possibilités actuelles de réduction des risques comportent aussi des risques. À la place, ces femmes peuvent choisir de recourir à une surveillance accrue, indépendamment de leur statut « BRCA ». Jusqu’à ce qu’une législation spécifique contre la discrimination génétique soit adoptée, les femmes doivent aussi tenir compte de la possibilité d’un usage discriminatoire des résultats de test. » (NBCC, 2001).
). Pour cette organisation, celle-ci est d’autant plus nécessaire que l’exemple des tests « BRCA » montre comment un service commercial de test a été mis en œuvre sans évaluation préalable de son intérêt alors même que ni la surveillance radiologique, ni la chirurgie prophylactique, ni la prévention hormonale ne constituent des formes d’intervention satisfaisante. Dans la dernière en date de ses prises de position, le groupe « génétique » de la NBCC concluait ainsi : « Un test génétique pour le cancer du sein ne doit pas être utilisé sans une prise en compte complète de ses limites et de ses implications. Les tests apportent peu de bénéfices. (…) S’ils peuvent aider les femmes ayant une importante histoire familiale de cancer du sein et qui sont porteuses de mutations des gènes BRCA 1/2 à prendre des décisions, il faut se souvenir que les possibilités actuelles de réduction des risques comportent aussi des risques. À la place, ces femmes peuvent choisir de recourir à une surveillance accrue, indépendamment de leur statut « BRCA ». Jusqu’à ce qu’une législation spécifique contre la discrimination génétique soit adoptée, les femmes doivent aussi tenir compte de la possibilité d’un usage discriminatoire des résultats de test. » (NBCC, 2001).Cette prudence à l’endroit des innovations n’est pas propre à la question des tests génétiques. La NBCC, comme la majorité des associations cancer du sein nord-américaines, a été fortement influencée par les formes d’action des malades atteints du sida, par leur capacité à intervenir non seulement dans les débats sur la prise en charge sociale mais aussi dans les débats scientifiques et techniques. Sans nier la nécessité d’un dialogue avec les experts officiels, la NBCC se voit comme une organisation indépendante qui exprime les opinions des malades et les représente face aux institutions biomédicales. Elle met l’accent sur une évaluation autonome des données, sur la confrontation entre les divers points de vue des experts, sur la diversité des pratiques de soin. Une telle évaluation, couplée avec une collecte systématique des avis des utilisateurs des techniques médicales, est perçue comme une condition indispensable à une véritable liberté de choix des malades. La NBCC a ainsi plaidé à la fois pour un recours limité aux tests, pour une intégration forte entre dépistage, conseil génétique et suivi des femmes dans le cadre de consultations spécialisées, et pour une régulation publique.
La discussion conduite par le SACGT est encore aujourd’hui l’expérience la plus approfondie de débat sur les contenus d’une régulation publique des tests génétiques. Dans ses conclusions, le SACGT insistait sur la spécificité des investigations génétiques. Le comité estimait que cette spécificité n’a pas tant pour origine le fait que les analyses génétiques auraient des caractéristiques uniques mais le fait qu’elles font intervenir une combinaison de problèmes particuliers. Ceux-ci tiennent à la nature d’une information qui constitue une identification de la personne, à l’horizon temporel de longue durée (parfois des dizaines d’années) des événements faisant l’objet de la prévision probabiliste, à l’implication directe de personnes autres que celui ou celle qui est à l’origine de l’analyse génétique, aux effets sociaux et psychologiques importants de certains tests.
Dans cette perspective, le comité a considéré que les risques posés par certaines catégories de test étaient de quatre ordres :
• des risques techniques (par exemple si les procédures ne permettent pas de détecter les changements génétiques recherchés et engendrent des faux négatifs, ou à l’inverse si elles détectent des modifications non pertinentes pour la survenue de la pathologie et produit des faux positifs) ;
• des risques cliniques (par exemple si le test est pratiqué de telle sorte qu’on effectue sur les personnes « à risque » des interventions comme moyen de contrôle ou de prévention non avérée ou douteuse) ;
• des risques iatrogènes (liés au fait que l’acquisition d’une information complexe et parfois très difficile à gérer peut engendrer suffisamment d’anxiété ou de désordres psychologiques pour aggraver l’état de la personne) ;
• des risques sociaux (liés par exemple aux pratiques de modulation des primes d’assurance ou de discrimination dans l’emploi).
En conséquence, le SACGT a pris en compte quatre cibles différentes pour la surveillance des tests, chaque cible requérant des mécanismes d’évaluation différents. La validité analytique, c’est-à-dire la capacité du test à détecter les modifications moléculaires qu’il est supposé détecter, est elle-même fonction de la précision et de la fiabilité des procédures.
La validité clinique est plus complexe. Elle renvoie à la relation entre les résultats de tests et la survenue d’une maladie. Son évaluation est affaire de sensibilité, de spécificité et de valeur prédictive, lesquelles varient considérablement suivant le type de pathologie, la pénétrance… La validité clinique dépend aussi de la prévalence de la maladie ainsi que des objectifs du test (diagnostic, pronostic ou prédictif).
L’utilité clinique renvoie à la possibilité d’une intervention préventive ou thérapeutique à la suite des analyses. En leur absence ou en l’absence de consensus sur l’efficacité de la prise en charge, le bénéfice se ramène à la possibilité de réduire l’incertitude, ce qui n’est pas automatiquement considéré comme un avantage. La question de l’utilité clinique fait aussi intervenir la nature du système de santé dans lequel s’inscrit la pratique du test : un test dont la validité clinique est parfaitement attestée peut dans un contexte défavorable (personnel non formé, offre mal organisée, absence de conseil à la fourniture des résultats) devenir contre-productif.
L’utilité sociale est la plus difficile à juger. Elle prend en compte à la fois les bénéfices des tests sur l’état de santé des populations, les problèmes d’égalité et de garantie d’accès aux services, l’impact économique mais aussi les risques de discrimination et de stigmatisation liés aux transformations du statut des personnes malades et/ou testées.
Le compromis finalement adopté puis proposé au gouvernement fédéral suppose l’implication de la FDA comme instance de régulation selon un dispositif proche de ceux adoptés pour les médicaments (SACGT, 2000).
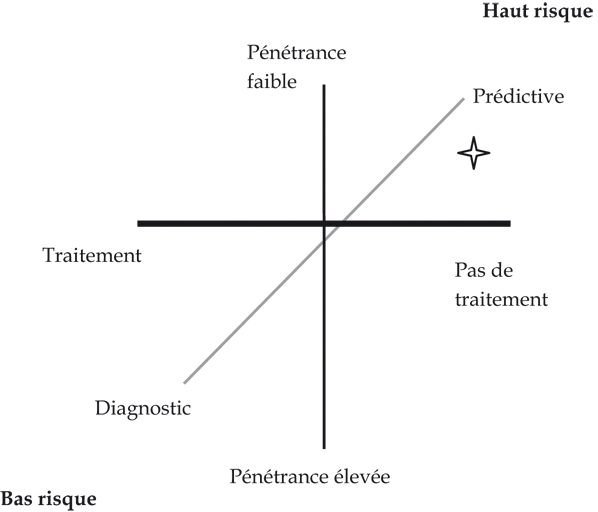
).Un élément essentiel de cette réflexion est la distinction entre les catégories de tests requérant ou non une surveillance étroite. Sans définir une grille formelle, le rapport final proposait une série de critères permettant de distinguer des tests à haut risque et des tests à bas risque (figure 10.1). Le classement envisagé reposait sur la prise en compte des paramètres suivants : détection de variations génétiques somatiques ou germinales, utilisation diagnostique ou prédictive, importance de la population cible, complexité des données et de leur interprétation, pénétrance des mutations, existence d’une intervention préventive ou curative de la maladie, utilisation potentielle sur des individus ou en population, risque de stigmatisation. Sur cette base, il existe un gradient d’impact des tests avec à un extrême la recherche de variants prédisposant à la maladie d’Alzheimer et à l’autre extrême la recherche des mutations du gène CF impliqué dans la mucoviscidose et entre les deux les tests « BRCA ». On peut accorder plus ou moins de poids à ces critères, le SACGT a par exemple proposé un schéma simple à trois dimensions : utilisation diagnostique ou prédictive, pénétrance et existence d’un traitement.
). Le classement envisagé reposait sur la prise en compte des paramètres suivants : détection de variations génétiques somatiques ou germinales, utilisation diagnostique ou prédictive, importance de la population cible, complexité des données et de leur interprétation, pénétrance des mutations, existence d’une intervention préventive ou curative de la maladie, utilisation potentielle sur des individus ou en population, risque de stigmatisation. Sur cette base, il existe un gradient d’impact des tests avec à un extrême la recherche de variants prédisposant à la maladie d’Alzheimer et à l’autre extrême la recherche des mutations du gène CF impliqué dans la mucoviscidose et entre les deux les tests « BRCA ». On peut accorder plus ou moins de poids à ces critères, le SACGT a par exemple proposé un schéma simple à trois dimensions : utilisation diagnostique ou prédictive, pénétrance et existence d’un traitement. | Figure 10.1 Schéma des différents critères permettant de distinguer des tests à haut risque et des tests à bas risque (d’après SACGT, 2000) |
Concrètement, le SACGT a suggéré la mise en place d’une procédure d’examen préalable à l’attribution d’un permis de commercialisation. Ce dispositif était destiné à compléter les procédures de surveillance existante : celle de la qualité technique des laboratoires et des procédures par le biais des accréditations (système dit CLIA) et la collecte d’informations épidémiologiques sur les mutations menée par les CDC. Le système de permis signifie que les développeurs de tests organiseraient le recueil des informations nécessaires à l’évaluation de la validité analytique et de l’utilité clinique des nouveaux tests. Parallèlement, le comité reprit la formule des autorisations temporaires inventée pour accélérer la recherche thérapeutique contre le sida. Le SACGT a proposé une phase de « pré-autorisation » pour ne pas freiner l’innovation et tenir compte de la difficulté à obtenir les données nécessaires au jugement des bénéfices cliniques. Dans la première phase, l’inventeur seul aurait la charge du recueil des données. Dans la seconde, les CDC avec la collaboration des praticiens seraient concernés puisqu’il s’agit de formes adaptées de surveillance post-marketing. Enfin, trait original, le comité évoqua la mise sur pied, indépendamment de la FDA, d’une évaluation éthique et sociale de l’impact des tests génétiques.
Adoptées comme position officielle de l’administration dans les derniers temps du gouvernement Clinton, les recommandations du SACGT sont restées lettre morte depuis la victoire des républicains à l’élection présidentielle de la fin 2000. Dans le contexte politique actuel, n’y a que très peu de chances que celles-ci soient traduites en réglementation par l’administration fédérale ou le Congrès.
Préserver la liberté de choix des consommateurs
En Grande-Bretagne, les cinq dernières années ont vu l’établissement de plusieurs structures chargées de la surveillance et du débat sur les pratiques de génétique humaine, parallèlement aux comités spécifiques des organismes professionnels et sociétés savantes. Il s’agit d’une part du Genetic Testing Network mis en place par le NHS pour coordonner l’offre de test et la standardisation des pratiques et d’autre part, depuis 1999, d’une commission permanente indépendante de l’administration sanitaire, la Human Genetics Commission (HGC) (NHS, 2003). Cette dernière a pris le relais d’un comité consultatif créé en 1997 et dispose d’un mandat large d’examen et d’écriture de recommandations.
). Cette dernière a pris le relais d’un comité consultatif créé en 1997 et dispose d’un mandat large d’examen et d’écriture de recommandations.En 2001, suite à une demande d’avis émanant d’une compagnie impliquée dans la fourniture de nutriments utiles à la santé alors désireuse de développer un service de tests pour les gènes impliqués dans les processus de détoxification, la Human Genetics Commission a lancé une réflexion sur l’offre de tests génétiques « directement au public » (HGC, 2002). Au-delà de cette incitation, la commission jugeait le problème préoccupant pour deux raisons. Premièrement, l’évolution très rapide des techniques de tests permet à la fois de réduire les coûts et d’élargir considérablement la palette des mutations recherchées. Deuxièmement, le cadre légal existant en Grande-Bretagne laisse totalement ouverte la possibilité d’une commercialisation directe, indépendamment de ce que fait ou ne fait pas le NHS.
). Au-delà de cette incitation, la commission jugeait le problème préoccupant pour deux raisons. Premièrement, l’évolution très rapide des techniques de tests permet à la fois de réduire les coûts et d’élargir considérablement la palette des mutations recherchées. Deuxièmement, le cadre légal existant en Grande-Bretagne laisse totalement ouverte la possibilité d’une commercialisation directe, indépendamment de ce que fait ou ne fait pas le NHS.Quoique le mandat de la HGC soit assez différent de celui du SACGT, la commission britannique remplit des fonctions proches de celles du comité américain. D’une part, elle assure une certaine représentation des acteurs et « intérêts » impliqués dans les pratiques de tests puisqu’elle inclut des experts (généticiens, cliniciens, médecins généralistes, juristes, bioéthiciens, industriels de la pharmacie), des membres provenant de l’administration du NHS et une personne issue d’association de personnes handicapées. D’autre part, la HGC entend pratiquer une certaine ouverture et transparence et cherche à impliquer les usagers et le « public » dans ses délibérations. Dans le cas du débat sur l’accès direct aux tests génétiques, cette ambition s’est traduite par la commande d’un sondage auprès de la population générale, et par l’organisation d’un travail avec des « profanes » dans le cadre d’ateliers scénarii (People Science and Policy, 2002).
).La HGC a retenu une définition large des tests génétiques dans laquelle sont incluses non seulement les analyses d’ADN mais aussi l’ensemble des procédures qui permettent une identification du patrimoine génétique d’une personne (HGC, 2003). L’existence d’une spécificité de ces procédures, même si elle est peu fondée d’un strict point de vue biologique, est reconnue importante en fonction de deux types de considérations. Premièrement, comme le SACGT américain, la HGC met en avant la capacité à prédire à long terme, la complexité de l’information produite par les tests et son caractère collectif. Deuxièmement, la commission considère que les tests génétiques sont de toute façon appréhendés comme spécifiques par la très grande majorité de la population et que cela justifie une gestion particulière.
). L’existence d’une spécificité de ces procédures, même si elle est peu fondée d’un strict point de vue biologique, est reconnue importante en fonction de deux types de considérations. Premièrement, comme le SACGT américain, la HGC met en avant la capacité à prédire à long terme, la complexité de l’information produite par les tests et son caractère collectif. Deuxièmement, la commission considère que les tests génétiques sont de toute façon appréhendés comme spécifiques par la très grande majorité de la population et que cela justifie une gestion particulière.Une seconde réflexion importante de la HGC porte sur ce qui doit être considéré comme un accès direct. Une définition large a été choisie pour tenir compte de la multiplicité des voies de commercialisation. Il ne s’agit donc pas uniquement des tests qui seraient mis à disposition sous la forme de kits utilisables « à la maison » (comme dans le cas des tests de grossesse) mais de l’ensemble des réseaux de fourniture ne passant pas par les procédures habituelles de référence médicale. L’ensemble des moyens de diffusion « hors NHS » est concerné, y compris la fourniture de tests par d’autres professionnels de santé que les médecins, par exemple par les pharmaciens, les infirmières, les psychologues ou les nutritionnistes.
La HGC a, dans un premier temps, entrepris un bilan de l’ensemble des règles et dispositifs constitutifs de la régulation britannique des tests (HGC, 2002). Par rapport à ce qui a été dit plus haut de la régulation étatique, le travail de la commission souligne que la fourniture de tests génétiques est, en Grande-Bretagne, une activité commerciale ouverte. La surveillance procède actuellement de trois horizons : les codes de conduite volontaires de l’industrie, les règles européennes, la réglementation britannique du commerce. Pour les premières, il n’y a aucun engagement particulier, sauf dans le cas des assurances avec la reconduction d’un moratoire volontaire. De leur côté, les directives européennes sur le diagnostic in vitro et sur les dispositifs médicaux officialisent l’obligation d’enregistrement et d’examen d’un dossier technique fourni par le développeur afin de prouver que le test détecte bien ce qu’il prétend détecter. Dans ce cadre, la surveillance des tests relèvera d’une nouvelle agence (Medical and Health Related-products Agency ou MHRA) mise en place pour unifier les institutions s’occupant d’une part des médicaments et d’autre part des dispositifs médicaux. Pour ce qui est de la réglementation commerciale, elle permet de contrôler la publicité même si les tests ne sont pas assimilés à des médicaments (pour lesquels la publicité directe est interdite) car les règles s’imposant à toute entreprise commerciale autorise l’engagement de procédures pour publicité mensongère. On se trouve donc dans une situation où seules les caractéristiques analytiques des tests font l’objet d’un examen extérieur au NHS.
). Par rapport à ce qui a été dit plus haut de la régulation étatique, le travail de la commission souligne que la fourniture de tests génétiques est, en Grande-Bretagne, une activité commerciale ouverte. La surveillance procède actuellement de trois horizons : les codes de conduite volontaires de l’industrie, les règles européennes, la réglementation britannique du commerce. Pour les premières, il n’y a aucun engagement particulier, sauf dans le cas des assurances avec la reconduction d’un moratoire volontaire. De leur côté, les directives européennes sur le diagnostic in vitro et sur les dispositifs médicaux officialisent l’obligation d’enregistrement et d’examen d’un dossier technique fourni par le développeur afin de prouver que le test détecte bien ce qu’il prétend détecter. Dans ce cadre, la surveillance des tests relèvera d’une nouvelle agence (Medical and Health Related-products Agency ou MHRA) mise en place pour unifier les institutions s’occupant d’une part des médicaments et d’autre part des dispositifs médicaux. Pour ce qui est de la réglementation commerciale, elle permet de contrôler la publicité même si les tests ne sont pas assimilés à des médicaments (pour lesquels la publicité directe est interdite) car les règles s’imposant à toute entreprise commerciale autorise l’engagement de procédures pour publicité mensongère. On se trouve donc dans une situation où seules les caractéristiques analytiques des tests font l’objet d’un examen extérieur au NHS.L’argument selon lequel l’accès à l’information génétique serait un droit des personnes a été comme tel refusé par la commission. En revanche, celle-ci a accordé une grande importance à l’idée selon laquelle les relations entre médecins et patients ont profondément évolué, qu’elles ne relèvent plus d’un modèle paternaliste, de sorte qu’il appartient au droit des patients de faire des choix, y compris en matière de demande de test. Pour la HGC, la pondération entre bénéfices et risques des tests est en dernière instance l’affaire des personnes. En fonction de cette liberté, on ne saurait interdire l’accès à telle ou telle catégorie de test même si on peut en encadrer les conditions (HGC, 2003). Estimant qu’il existe en Grande-Bretagne un consensus (le sondage réalisé auprès de la population générale va dans ce sens) pour considérer que le NHS doit rester le pourvoyeur et le gestionnaire de l’information génétique médicale, la principale crainte que la commission entend prendre en compte est le possible usage discriminatoire de cette information. En pratique, cela signifie que les cibles d’une régulation supplémentaire des tests ne portent ni sur la validité analytique pour laquelle le cadre existant est considéré comme satisfaisant ni sur l’utilité clinique qui est l’affaire des experts du NHS mais sur les usages sociaux plus larges des résultats de test.
). Estimant qu’il existe en Grande-Bretagne un consensus (le sondage réalisé auprès de la population générale va dans ce sens) pour considérer que le NHS doit rester le pourvoyeur et le gestionnaire de l’information génétique médicale, la principale crainte que la commission entend prendre en compte est le possible usage discriminatoire de cette information. En pratique, cela signifie que les cibles d’une régulation supplémentaire des tests ne portent ni sur la validité analytique pour laquelle le cadre existant est considéré comme satisfaisant ni sur l’utilité clinique qui est l’affaire des experts du NHS mais sur les usages sociaux plus larges des résultats de test.Concrètement, la HGC a repris les catégories analytiques élaborées par le SACGT pour réfléchir à des scénarii alternatifs allant de l’absence de régulation de l’offre commerciale à la généralisation d’un statut équivalent à celui du médicament avec une obligation de prescription (HGC, 2003). Le schéma retenu dans les recommandations (soutenu par la majorité des personnes auditionnées et consultées) consiste en une régulation partielle dans laquelle une partie des tests seront à prescription obligatoire et d’autres en accès direct. Les critères de classement sont renvoyés d’une part à la discussion du SACGT, d’autre part à une réflexion ultérieure. Le principal paramètre évoqué est la complexité de la maladie et l’incertitude des prédictions dans le cas des maladies multifactorielles. La HGC a ainsi écarté le mécanisme proposé par les organisations de consommateurs qui était une généralisation du dispositif existant en Grande-Bretagne pour les tests VIH, c’est-à-dire une combinaison entre un examen préalable de la qualité et la prescription obligatoire.
). Le schéma retenu dans les recommandations (soutenu par la majorité des personnes auditionnées et consultées) consiste en une régulation partielle dans laquelle une partie des tests seront à prescription obligatoire et d’autres en accès direct. Les critères de classement sont renvoyés d’une part à la discussion du SACGT, d’autre part à une réflexion ultérieure. Le principal paramètre évoqué est la complexité de la maladie et l’incertitude des prédictions dans le cas des maladies multifactorielles. La HGC a ainsi écarté le mécanisme proposé par les organisations de consommateurs qui était une généralisation du dispositif existant en Grande-Bretagne pour les tests VIH, c’est-à-dire une combinaison entre un examen préalable de la qualité et la prescription obligatoire.Compte tenu des nouvelles possibilités technologiques, le HGC considère toutefois qu’il est préférable de ne pas développer des tests utilisables hors contexte professionnel (home kits). Il faut donc une nouvelle régulation. Celle-ci est renvoyée à l’activité de deux instances déjà existantes. D’une part, la future MHRA se chargera de tout ce qui est de l’homologation et de la qualité technique. D’autre part, compte tenu du peu de développement envisagé pour les pratiques de test hors du NHS, la question de l’utilité clinique est renvoyée à ce dernier. L’évaluation de l’utilité resterait de l’ordre des compétences du Genetic Testing Network mis en place au sein du NHS. C’est ce réseau de spécialistes qui est, pour la commission, le mieux placé pour faire la pondération risques/bénéfices et réfléchir aux conditions d’accès et d’usage. Dans ce cas, rien ne changerait par rapport aux formes de régulation professionnelle internes au système de santé publique. Les recommandations manifestent toutefois une incertitude sur ce point dans la mesure où pour prolonger sa comparaison entre tests et médicaments revendiqués, la commission estime que les missions de la MHRA devraient aussi inclure un examen de l’utilité clinique (HGC, 2003).
).Le débat britannique sur l’accès direct manifeste donc de fortes tensions entre deux approches de la régulation. La première est proche des cadres industriel et consumériste avec l’insistance sur la liberté des usagers et la dimension individuelle des jugements risques/bénéfices. La seconde approche est celle d’une régulation étatique maintenue et éventuellement renforcée dans laquelle le réseau NHS permet de contenir l’expansion des marchés et du même coup limite le risque de voir des tests inutiles ou de qualité insuffisante mis à disposition des généralistes et du public. À l’inverse de ce qui s’était passé dans le débat américain, la question de l’utilité et du même coup les dimensions pratiques de la régulation ont été largement délaissées par la HGC. Ses recommandations laissent ouverte la possibilité de développements de service de test hors du cadre NHS comme par exemple c’est le cas avec les premières offres de diagnostic préimplantatoire des mutations BRCA. Mais la commission estime qu’il s’agit-là de phénomènes marginaux.
Orientations possibles pour la surveillance et la régulation des tests génétiques
Cette présentation de quelques-uns des débats qui ont, à l’étranger, porté sur la régulation des tests génétiques n’est pas un état des connaissances à proprement parler. Elle permet cependant de formuler un certain nombre de conclusions qui sont autant de points de référence pour la définition d’une politique de régulation adaptée au système de santé français.
Les tests génétiques (entendus ici comme toutes les explorations donnant accès au patrimoine héréditaire d’un individu ou d’une famille, qu’il s’agisse ou non d’analyse de la structure moléculaire de l’ADN) sont des outils complexes. Leur validité ne repose pas sur une relation simple et univoque entre génotype et phénotype, entre génotype et pathologie. Le concept de « déterminisme génétique » est, de ce point de vue, malheureusement source de nombreuses confusions, en particulier lorsqu’il est employé pour définir des politiques sanitaires.
Cette complexité biologique et médicale n’est pas qu’un enjeu de recherche ou de connaissance. Elle caractérise aussi les conditions d’usage des tests et l’évaluation de leurs impacts psychologiques, sociaux et culturels. Face à une vision naïvement optimiste des innovations biomédicales, il est bon de se souvenir que l’information n’est pas bonne en soi ; son sens et ses effets dépendent dans une très large mesure des contextes dans lesquels elle est produite et délivrée, des interventions auxquelles elle est associée, des acteurs qui s’en saisissent. De même, les tests génétiques peuvent aussi avoir des effets négatifs directs (par exemple en matière de discrimination à l’emploi ou à l’assurance) ou indirects (par exemple à cause des effets psychologiques anxiogènes de la connaissance d’un risque de maladie).
Par rapport aux autres outils de la biomédecine, les tests génétiques présentent une spécificité relative. Celle-ci tient tout d’abord à la nature de l’information apportée par les tests : le plus souvent un risque de maladie et une modalité de transmission et pas seulement le diagnostic d’un état pathologique. La prédiction associée au test peut parfois porter sur des événements à plusieurs dizaines d’années de distance. Elle tient ensuite aux implications médicales de cette mise en évidence. Les résultats de test concernent directement à la fois un individu et un collectif même si les membres de ce dernier ne se sont pas associés ni même informés de la démarche de test. Le statut particulier des tests est renforcé par le « gap médical » qui caractérise aujourd’hui de nombreuses pathologies pour lesquelles nous disposons de grande capacité d’identification des facteurs de risque sans moyens d’intervention préventive ou thérapeutique efficaces.
En conséquence, les tests génétiques posent des problèmes particuliers d’évaluation et de régulation. L’enjeu n’est pas ici la question de la qualité technique (validité analytique) des tests pour lesquels les mécanismes d’accréditation et de validation « interne » à l’exercice de la génétique médicale peuvent être jugés satisfaisants mais la garantie d’utilité médicale et sociale des tests.
En conclusion, la régulation des tests pose des problèmes d’accès et d’équité dans cet accès. Elle pose aussi des problèmes de régulation des marchés ou de contrôle du « risque commercial ». Celui-ci correspondant à la possibilité pour un opérateur de monopoliser l’usage des gènes et la réalisation des tests qui amplifient les coûts et limitent l’innovation, comme l’a montré la trajectoire des tests « BRCA ». Du fait des relations étroites qui peuvent lier opérateurs commerciaux, prescripteurs et parfois (publicité directe) les patients ou leurs familles, il existe aussi un risque d’excès de tests, c’est-à-dire de développement de pratiques de tests médicalement peu ou pas « utiles ».
Dans ce contexte, il semble qu’une initiative en matière de régulation devrait s’appuyer sur les orientations suivantes. Il faut conserver l’approche intégrée évitant une séparation entre réalisation technique des tests et utilisation clinique, il ne faut donc pas s’engager dans la voie du « laboratoire d’analyse génétique » sur le modèle du laboratoire d’analyses biologiques autonome. La régulation doit porter sur plusieurs niveaux : validité analytique, spécificité analytique, utilité clinique et impact social ; elle ne peut donc pas faire intervenir un dispositif unique. Tous les tests ne posent pas les mêmes problèmes et ne devraient pas être soumis aux mêmes exigences. La mise en place d’une régulation publique doit impliquer les praticiens, les administrations sanitaires, les chercheurs mais aussi les patients et les familles selon des modalités ouvertes de représentation. Une régulation efficace implique la mise en place d’une procédure d’évaluation de l’utilité des tests préalable à leur introduction en routine, que celle-ci se fasse par le biais d’une mise sur le marché, d’une accréditation de laboratoires ou de l’instauration d’une procédure de remboursement. La mise en place de cette évaluation préalable requiert d’approfondir la discussion publique sur les critères de jugement, les dispositifs de collecte de l’information, les modalités de prise de décision.
Bibliographie
[1]american society of clinical oncology (asco). Genetic Testing for Cancer Susceptibility.
Journal of Clinical Oncology. 1996;
14:1730- 1736
[2] angell m. The Truth About Drug Companies.
How They Deceive Us and What to Do About It. New York:Random House;
2004;
[3] aronowitz r. Making Sense of Illness : Science, Society and Disease.
Cambridge University Press;
Cambridge:1998;
[4] barbot j. Les malades en mouvement.
Balland;
Paris:2002;
[5] baszanger i, gaudillière jp, löwy i. Légitimer et réguler les innovations biomédicales.
Sciences Sociales et Santé. 2000;
18:
[6] berlivet l. Une santé à risque. Thèse.
Université de Rennes. 2000;
[7] bonah c, rasmussen a. Histoire et Médicament aux 19e et 20e siècles, Biotem et Glyphe.
Paris:2005;
[8] bourret p. BRCA Patients and Clinical Collectives: New Configurations of Action in Cancer Genetic Practices. ».
Social Studies of Science. 2005;
35:41- 68
[9] callon m. Four models of the dynamics of science ».
In : Handbook of Science and Technology Study. In: jasanoff s (ed), editors.
1995;
[10] callon m, rabeharisoa v. Le pouvoir des malades.
Éditions de l’École des Mines;
Paris:1999;
[11] callon m, lascoumes p, barthes y. Agir dans un monde incertain. Essai sur la démocratie technique.
Seuil;
Paris:2001;
[12] cambrosio a, keating p. Biomedical Platforms.
MIT Press;
Cambridge:2003;
[13] casamayou mh. The Politics of Breast Cancer.
Georgetown University Press;
Washington:2001;
[14] chauveau s. L’invention pharmaceutique. La pharmacie française entre l’État et la société au 20e siècle. Les Empêcheurs de Penser en Rond.
Paris:1999;
[15] chauveau s. Industries du médicament et du vivant.
Entreprise et histoire. 2004;
36n° spécial:
[16] cooter r, pickstone j. Medicine in the twentieth century.
Harwood;
Amsterdam:2000;
[17] coriat b. Les droits de propriété intellectuelle nouveaux domaines, nouveaux enjeux.
La Revue d’Economie Industrielle. 2002;
99n° spécial:
[18] daemmerich a. Pharmacopolitics. Drug Regulation in the United States and Germany.
Chapel Hil:University of North Carolina Press;
2004;
[19] dalgalarondo s. Sida : La course aux molécules.
Éditions EHESS;
Paris:2003;
[20] dasgupta p, david p. Towards a new economics of science.
Research Policy. 1994;
23:487- 521
[21] dodier n. Leçons politiques de l’épidémie de Sida.
Éditions EHESS;
Paris:2003;
[22] donai d, rob e. Integrated Regional Genetic Services: Current and Future Provision.
British Medical Journal. 2001;
322:1048- 1052
[23] douglas m. Risk and acceptability according to the Social Sciences.
Basic Book;
New York:1986;
[24] eisenberg r. The move towards the privatization of biomedical research.
In : The Future of Biomedical Research. In: barfield ce, smith lr (eds), editors.
AEI Press;
New York:1997;
[25] epstein s. Impure Science.
University of California Press;
Berkeley:1996;
[26] etzkowitz h, leydesdorff l. Universities and the global knowledge economy. A triple helix of university-industry-government.
Pinter;
London:1997;
[27] foray d. Science, Technology and the Market.
World Science Report, UNESCO. 1999;
[28] gaudillière jp. Vers une nouvelle régulation du médicament ? Les thérapies hormonales substitutives entre profession, État, marché et usagers, 1930-2005.
In : La gouvernance des innovations médicales. In: tournay v (coord.), editors.
PUF. 2007;
257276
[29] gaudillière jp, cassier m. Production, valorisation et usage des savoirs : la génétique du cancer du sein.
MIRE;
Paris:2001;
[30] gaudillière jp, löwy i. Medicine, markets and public health: Contemporary testing for breast cancer predispositions.
In : Medicine, markets and mass media: Producing health in the twentieth century. In: berridge v, loughin k (eds), editors.
London:Routledge;
2005;
[31] gibbons m, limoges c, nowotny h, schwartzman s, scott p, trow m. The New Mode of Knowledge Production.
Sage;
London:1994;
[32] gilbert c. Risques collectifs et situation de crise. Apports de la recherche en sciences humaines et sociales.
L’Harmattan;
Paris:2003;
[33] goozner m. The 800 Million Dollars Pill. The Truth Behind the Cost of New Drug.
University of California Press;
Berkeley:2004;
[34] holtzmann n, watson s. Promoting Safe and Effective Genetic Testing in the United States: Final Report of the Task Force on Genetic Testing.
The Johns Hopkins University Press;
Baltimore:1998;
[35]human genetics commission (hgc). The supply of genetic tests direct to the public.
A consultation document. 2002;
Site Internet : www.hgc.gov.uk.
[36]human genetics commission (hgc). Genes Direct. Ensuring the effective over-sight of genetic tests supplied directly to the public.
London, Department of Health:2003;
[37]inserm, fnclcc. Risques héréditaires des cancers du sein et de l’ovaire. Quelle prise en charge?.
Collections Expertise Collective. Éditions Inserm;
Paris:1998;
[38] joly pb. Les OGM entre la science et le public. Quatre modèles pour la gouvernance de l’innovation et des risques.
Economie rurale. 2001;
266:11- 29
[39] julian-reynier c, pierret j, eisinger f. Prédisposition génétique aux cancers : questions psychologiques et débats de société.
John Libbey;
Paris:2005;
[40] löwy i. Between Bench and Bedside.
Harvard University Press;
Cambridge:1996;
[41] marks h. The Progress of Experiment. Science and Therapeutic Reform in the United States.
Cambridge University Press;
Cambridge. 1997;
[42] mirowski p. Science Bought and Sold. Essays in the Economics of Science.
University of Chicago Press;
Chicago:2002;
[43]national breast cancer coalition (nbcc). Genetic Testing for Heritable Breast Cancer Risk.
Position paper. 2001;
Site Internet : www.nbcc.org.
[44]national health service (nhs). Our Inheritance, our future. Realising the potential of genetics in the NHS.
Department of Health, London:2003;
[45] parthasarathy s. Architectures of Genetic Medicine. Comparing Genetic Testing for Breast Cancer in the USA and the UK.
Social Studies of Science. 2005;
35:5- 40
[46]people science and policy ltd. The supply of genetic tests direct to the public. A report prepared for the Human Genetics Commission.
London:December 2002;
Site Internet : www.peoplescienceandpolicy.com.
[47] pestre d. Science, argent et politique. Un essai d’interprétation.
INRA Éditions;
Paris:2003;
[48] pignarre p. Le grand secret de l’industrie pharmaceutique.
La Découverte;
Paris:2003;
[49]secretary’s advisory committee on genetic testing. Report on Genetic Testing for Late Onset Disorders.
London: Health Department of the United Kingdom.
1998;
[50]secretary’s advisory committee on genetic testing, enhancing the over-signt of genetic tests (sacgt). Recommendations of the SACGT.
Washington, NIH:2000;
[51] sinding c. Le clinicien et le chercheur.
PUF;
Paris:1991;
[52]stanford program in genomics, ethics and society (spges). Genetic Testing for BRCA1 and BRCA2.
Journal of Women’s Health. 1998;
7:531- 545
[53] urfalino p. Le grand méchant loup pharmaceutique. Angoisse ou vigilance ?.
Textuel;
Paris:2005;
→ Aller vers SYNTHESE