Cancer du poumon
2008
| ANALYSE |
3-
Classification histologique et pathologie moléculaire
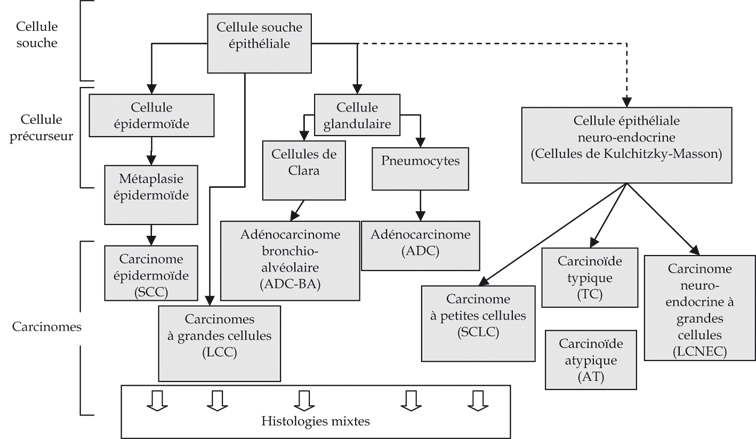
La plupart des cancers du poumon sont des carcinomes (les autres types histologiques représentent moins de 1 % des cas). Ces carcinomes se développent à partir de l’épithélium bronchique des voies respiratoires larges et moyennes, et des alvéoles pulmonaires (figure 3.1 ).
).
).
Histologie
Les cancers du poumon sont classés en deux grandes catégories : les carcinomes dits « non-à petites cellules » (Non-Small Cell Lung Carcinomas, NSCLC), qui dérivent des cellules souches épithéliales de la muqueuse broncho-pulmonaire, et les carcinomes dits « à petites cellules » (Small Cell Lung Carcinoma, SCLC) qui regroupent plusieurs catégories de cancers présentant des caractéristiques morphologiques, histologiques et ultrastructurales communes, dont en particulier la présence de granules neurosécréteurs et une importante activité mitotique.
Les NSCLC représentent 80 % des cas, et peuvent adopter une architecture épidermoïde (Squamous Cell Carcinoma, SCC), glandulaire (AdenoCarcinoma, ADC) ou indifférenciée (Large Cell Carcinoma, LCC), selon l’étiologie et la localisation dans l’arbre bronchique. L’histologie de l’arbre bronchique et pulmonaire se caractérise par le passage progressif d’un type d’épithélium à l’autre au fur et à mesure des ramifications. L’épithélium de type trachéal, pseudo-stratifié, cilié et contenant des cellules caliciformes, devient de moins en moins haut dans les bronches segmentaires pour céder la place, dans les bronchioles terminales, à un épithélium cylindrique simple, dépourvu de cellules caliciformes et caractérisé par un type cellulaire spécialisé, la cellule de Clara. Les bronchioles terminales s’ouvrent sur les alvéoles pulmonaires, tapissées de pneumocytes de type I et II. Le SCC se développe à partir de l’épithélium bronchique pseudo-stratifié par un processus de métaplasie épidermoïde, suivi d’une séquence hyperplasie-dysplasie-carcinome. Les carcinomes broncho-alvéolaires se développent principalement à partir de la muqueuse à cellules de Clara des petites bronchioles périphériques. La transformation des pneumocytes de type I et II donne naissance à des adénocarcinomes. La proportion des différents types histologiques diffère en fonction du sexe et de l’exposition au tabac. Les SCC représentent 44 % des cancers du poumon chez l’homme et 25 % chez la femme, et sont le type dominant de cancers chez les gros fumeurs. Les chiffres sont inverses pour les ADC, qui représentent 28 % des cancers chez l’homme et 42 % chez la femme.
L’origine, la diversité et la typologie des tumeurs neuro-endocrines restent un sujet de débat. On pense que ces tumeurs dérivent de cellules-précurseurs spécifiques présentant des caractéristiques neuro-endocrines (cellules de Kulchitzky-Masson). Au sens strict, le terme SCLC ne s’applique qu’à une catégorie de tumeurs dont les cellules sont pauvres en cytoplasme, avec une chromatine d’apparence granuleuse, une activité mitotique élevée et de grandes plages de nécrose. Les autres types de tumeurs neuro-endocrines sont les carcinomes neuro-endocrines à grandes cellules (Large Cell NeuroEndocrine Carcinomas, LCNEC), les carcinoïdes typiques (Typical Carcinoïd, TC) et les carcinoïdes atypiques (Atypical Carcinoïds, AT). Ces deux derniers types se distinguent par leur activité mitotique (plus faible pour les TC que pour les AT). Les carcinomes neuro-endocrines sont fortement associés au tabagisme, ont une croissance très rapide, une bonne réponse initiale à la chimiothérapie (conséquence probable de leur activité mitotique élevée) et une tendance très marquée à la formation de métastases.
Les cancers du poumon sont souvent hétérogènes sur le plan histologique, avec des variations d’apparence d’un champ microscopique à l’autre. On distingue des entités hybrides tels que des carcinomes adénosquameux et des carcinomes pleïomorphes. On trouve aussi des structures typiques de différenciation neuro-endocrine dans 10 à 20 % des SCC, ADC et LCC. Sur le plan moléculaire, ces tumeurs hybrides apparaissent comme étant clonales : on pense donc qu’elles dérivent de l’expansion d’une seule cellule transformée. Cette observation illustre le caractère plastique de la différenciation des cancers du poumon, ainsi que l’absence de frontières nettes entre les différents types histologiques.
Cancérogenèse moléculaire
Les cancers broncho-pulmonaires se développent selon un processus multi-étapes, caractérisé par une progression vers le phénotype invasif d’une ou d’un petit nombre de cellules « initiées » par l’acquisition d’altérations génétiques leur conférant un avantage prolifératif (Hanahan et Weinberg, 2000). De nombreux agents cancérogènes professionnels ou environnementaux, comme ceux présents dans la fumée du tabac, peuvent induire l’initiation des cellules bronchiques ou alvéolaires et favoriser leur progression. Ces agents affectent souvent l’arbre broncho-pulmonaire dans son ensemble (ainsi que, dans le cas de la fumée du tabac, l’ensemble des voies aéro-digestives supérieures) et peuvent « initier » de façon indépendante des cellules distantes les unes des autres, donnant naissance à plusieurs lésions primaires concomitantes. Ce phénomène est décrit sous le nom de « cancérogenèse de champ » (Field Carcinogenesis).
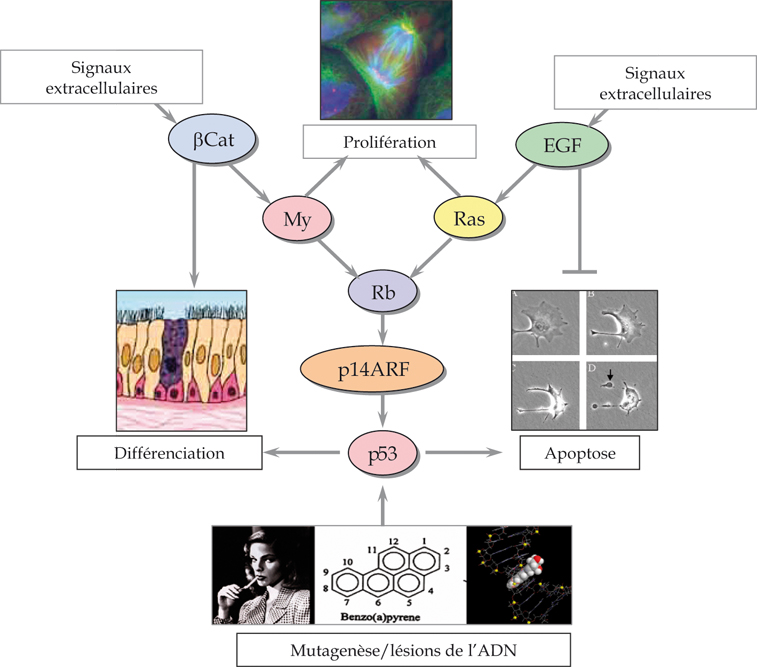
). De nombreux agents cancérogènes professionnels ou environnementaux, comme ceux présents dans la fumée du tabac, peuvent induire l’initiation des cellules bronchiques ou alvéolaires et favoriser leur progression. Ces agents affectent souvent l’arbre broncho-pulmonaire dans son ensemble (ainsi que, dans le cas de la fumée du tabac, l’ensemble des voies aéro-digestives supérieures) et peuvent « initier » de façon indépendante des cellules distantes les unes des autres, donnant naissance à plusieurs lésions primaires concomitantes. Ce phénomène est décrit sous le nom de « cancérogenèse de champ » (Field Carcinogenesis).Comme dans la plupart des cancers, les carcinomes broncho-pulmonaires acquièrent au cours de leur développement une variété d’altérations génétiques (mutations, amplifications géniques, pertes d’allèles, instabilités chromosomiques) et épigénétiques (surexpression des gènes, extinction de l’expression par hyperméthylation des promoteurs) (Yokota et Kohno, 2004). La fréquence et le type des altérations diffèrent d’une histologie à l’autre (figure 3.2). Cependant, quel que soit le type histologique, les mêmes voies sont souvent affectées par des mécanismes différents. On peut donc proposer que ces voies jouent un rôle fondamental dans la morphogenèse, les réponses au stress et la régénération après lésion de l’épithélium broncho-pulmonaire normal, définissant un « carrefour régulatoire » qui intègre prolifération, apoptose, différenciation et réponses aux lésions de l’ADN (figure 3.3). La conséquence biologique principale de ces altérations est de découpler ces mécanismes les uns des autres. Dès lors, la cellule affectée devient capable de proliférer au-delà de sa limite réplicative normale, de se maintenir en vie dans des conditions où la physiologie normale entraîne une mort cellulaire, d’éviter l’engagement dans les voies de différenciation terminale, et de se développer selon des schémas de différenciation altérés. Ces effets ne sont pas propres aux carcinomes broncho-pulmonaires : les mécanismes en question sont impliqués de façon très générale dans tous les types de cancers épithéliaux. Ce qui fait la particularité des carcinomes broncho-pulmonaires tient à une double caractéristique : la plasticité histologique de l’épithélium broncho-pulmonaire, décrite plus haut, qui confère à cet épithélium une forme d’instabilité tissulaire s’exprimant par la formation fréquente de métaplasies, et le poids particulier des facteurs de risque environnementaux, notamment de la fumée du tabac, qui agissent non seulement comme mutagènes mais aussi comme facteurs de remodelage de l’épithélium bronchique.
). La fréquence et le type des altérations diffèrent d’une histologie à l’autre (figure 3.2). Cependant, quel que soit le type histologique, les mêmes voies sont souvent affectées par des mécanismes différents. On peut donc proposer que ces voies jouent un rôle fondamental dans la morphogenèse, les réponses au stress et la régénération après lésion de l’épithélium broncho-pulmonaire normal, définissant un « carrefour régulatoire » qui intègre prolifération, apoptose, différenciation et réponses aux lésions de l’ADN (figure 3.3). La conséquence biologique principale de ces altérations est de découpler ces mécanismes les uns des autres. Dès lors, la cellule affectée devient capable de proliférer au-delà de sa limite réplicative normale, de se maintenir en vie dans des conditions où la physiologie normale entraîne une mort cellulaire, d’éviter l’engagement dans les voies de différenciation terminale, et de se développer selon des schémas de différenciation altérés. Ces effets ne sont pas propres aux carcinomes broncho-pulmonaires : les mécanismes en question sont impliqués de façon très générale dans tous les types de cancers épithéliaux. Ce qui fait la particularité des carcinomes broncho-pulmonaires tient à une double caractéristique : la plasticité histologique de l’épithélium broncho-pulmonaire, décrite plus haut, qui confère à cet épithélium une forme d’instabilité tissulaire s’exprimant par la formation fréquente de métaplasies, et le poids particulier des facteurs de risque environnementaux, notamment de la fumée du tabac, qui agissent non seulement comme mutagènes mais aussi comme facteurs de remodelage de l’épithélium bronchique.

Mutations de TP53
L’altération génétique la plus fréquente est la mutation du gène suppresseur TP53 (chromosome 17p13). Son produit, la protéine p53, est un facteur de transcription apparenté à une famille de protéines essentielles à la différenciation et à la morphogenèse épithéliale, mais spécialisé dans la réponse à un large spectre de stress physiques, chimiques ou biochimiques. P53 est un médiateur essentiel de la réponse des cellules aux expositions à des agents cancérogènes, capable d’entraîner l’arrêt du cycle cellulaire, la réparation de l’ADN ou l’apoptose en fonction du type cellulaire, du degré de différenciation, de la nature et de l’intensité du stress. Cette protéine occupe une position centrale dans le « carrefour de régulation » décrit plus haut. Son rôle de « capteur » des modifications environnementales en fait un acteur de premier plan dans la régulation de la stabilité génétique et tissulaire de l’épithélium broncho-pulmonaire.
Les mutations de TP53 sont principalement des substitutions faux-sens qui inactivent la protéine en empêchant son repliement dans une conformation active (Pfeifer et coll., 2002). On détecte des mutations de TP53 dans 50 % des NSCLC et dans plus de 70 % des SCLC.
). On détecte des mutations de TP53 dans 50 % des NSCLC et dans plus de 70 % des SCLC.Dans les SCC des gros fumeurs, la fréquence des mutations peut dépasser 80 %. Chez ces derniers, on retrouve des mutations dans les métaplasies ainsi que dans l’épithélium non pathologique : la mutation précède, en quelque sorte, la formation de la tumeur. En revanche, dans les ADC des femmes non-fumeuses, les fréquences décrites dans la littérature varient entre 25 et 50 % et on pense que ces mutations apparaissent à un stade plus tardif de la progression tumorale. Chez les fumeurs, la nature chimique de la mutation constitue souvent une « signature moléculaire » des agents mutagènes de la fumée du tabac, tels que le benzo(a)pyrène et d’autres hydrocarbures polycycliques aromatiques (Le Calvez et coll., 2005). Dans la cellule exposée, ces agents subissent une bio-activation qui génère des métabolites capables de se fixer sur l’ADN de façon covalente. Les métabolites du benzo(a)pyrène se fixent préférentiellement sur certaines guanines, et ces mêmes guanines sont fréquemment mutées dans les cancers des fumeurs. Cette « signature » moléculaire n’est pas présente dans les cancers des non-fumeurs.
). Dans la cellule exposée, ces agents subissent une bio-activation qui génère des métabolites capables de se fixer sur l’ADN de façon covalente. Les métabolites du benzo(a)pyrène se fixent préférentiellement sur certaines guanines, et ces mêmes guanines sont fréquemment mutées dans les cancers des fumeurs. Cette « signature » moléculaire n’est pas présente dans les cancers des non-fumeurs.En dépit de nombreux travaux, l’impact de la mutation de TP53 sur le pronostic et sur la prédiction des réponses thérapeutiques n’a pas été évalué de manière satisfaisante. Vu l’hétérogénéité des mutations et la diversité des cancers, la plupart des études menées à ce jour manquent de puissance statistique pour tirer des conclusions significatives.
Dérégulation de TP63 et TP73
Les deux autres membres de la famille TP53, TP63 et TP73, sont exprimés de façon complexe au cours de la morphogenèse et de la différenciation de l’arbre bronchique. Malgré leur ressemblance structurale et biochimique avec TP53, ces deux gènes ne sont pas des suppresseurs de tumeurs typiques. Ils sont néanmoins impliqués, au moins comme co-facteurs, dans la carcinogenèse bronchique. La protéine p63 est un facteur décisif dans la différenciation épidermoïde et son expression est indispensable à la formation de l’épithélium pluri-stratifié. Cette protéine est surexprimée (parfois en conséquence de l’amplification du gène, localisé en 3q28) dans les métaplasies de la muqueuse bronchique et dans tous les SCC. Elle constitue un bon marqueur histologique du compartiment épidermoïde des tumeurs présentant une histologie mixte. Le rôle de p73 est moins bien compris : cette protéine joue un double rôle dans la différenciation et la réponse au stress de nombreux types cellulaires (différenciation neuronale, épithéliale). Il est possible que certaines formes mutées de p53 interfèrent avec les protéines p63 et/ou p73, modifiant leurs activités. Cette interaction pourrait être à la base d’un effet pro-oncogénique (« gain-de-fonction ») de certains mutants, observé expérimentalement, mais dont la signification physiopathologique reste un sujet de débat.
Altérations de EGFR
Les altérations du récepteur de l’EGF (Epidermal Growth Factor) – EGFR –, aussi décrit sous les acronymes HER1 et ERBB1, sont fréquentes dans les adénocarcinomes, en particulier chez les non-fumeurs. Ce récepteur transmembranaire contient un domaine tyrosine-kinase intracellulaire et est stimulé par une famille de ligands comprenant, entre autres, l’EGF, le TGF-alpha (Transforming Growth Factor alpha), l’amphiréguline, l’épiréguline, et la betacelluline. Il appartient à une famille de quatre récepteurs de structure et de fonction apparentées (HER 1 à 4). Leur activation induit une cascade de transduction du signal modulant la prolifération, la survie, l’adhésion, la migration, la différenciation des cellules épithéliales et l’angiogenèse. Sur le plan moléculaire, la fixation du ligand entraîne la dimérisation des récepteurs (y compris la formation d’hétérodimères avec d’autres membres de la famille HER), l’activation de la tyrosine kinase et l’auto-phosphorylation de résidus tyrosine des récepteurs dimérisés. Ces phosphotyrosines constituent des sites de liaison pour des molécules de transduction du signal intracellulaire. Le signal de prolifération cellulaire est principalement dépendant du recrutement de complexes entre les protéines adaptatrices Grb2 et Sos qui fournissent une connexion avec les protéines de la famille Ras et la cascade des RAF/MAP kinases. Les effets anti-apoptotiques favorisant la survie cellulaire sont médiés, entre autres, à travers l’activation de la kinase cellulaire Akt (ou protéine kinase B).
Des mutations de l’EGFR sont détectables dans près de 50 % des ADC chez les sujets non-fumeurs (Shigematsu et Gazdar, 2006). À ce jour, la littérature mondiale fait état de 2 500 tumeurs analysées, et porte sur près de 500 mutations détectées. Ces mutations sont d’origine somatique et apparaissent en des sites précis des exons 18 à 21. Les mutations les plus fréquentes (représentant plus de 80 % de toutes les mutations décrites) sont des délétions de 2 à 6 codons dans l’exon 19, conservant le cadre de lecture, et la substitution d’une arginine en leucine au codon 858 dans l’exon 21. Des mutations ponctuelles dans l’exon 18 (codon 179) et des insertions dans l’exon 20, sont plus rarement observées. Ces différentes mutations affectent la structure de boucles protéiques encadrant le domaine de liaison de l’ATP qui constitue le site actif de l’enzyme (Lynch et coll., 2004). Elles entraînent une activation constitutive de la kinase, avec des différences d’effet en fonction de la nature et de la position de la mutation. D’autres modes d’activation oncogénique sont également décrits, tels que l’amplification génique ou la surexpression. Il semble que la mutation de l’EGFR soit un événement précoce dans la cancérogenèse chez les non-fumeurs. Les mécanismes de mutagenèse responsables de ces mutations ne sont pas connus, on sait cependant que les mutations de l’EGFR sont associées à l’altération de voies de sauvegarde et de maintien de l’intégrité génomique dont la voie p53 (Mounawar et coll., 2007).
). À ce jour, la littérature mondiale fait état de 2 500 tumeurs analysées, et porte sur près de 500 mutations détectées. Ces mutations sont d’origine somatique et apparaissent en des sites précis des exons 18 à 21. Les mutations les plus fréquentes (représentant plus de 80 % de toutes les mutations décrites) sont des délétions de 2 à 6 codons dans l’exon 19, conservant le cadre de lecture, et la substitution d’une arginine en leucine au codon 858 dans l’exon 21. Des mutations ponctuelles dans l’exon 18 (codon 179) et des insertions dans l’exon 20, sont plus rarement observées. Ces différentes mutations affectent la structure de boucles protéiques encadrant le domaine de liaison de l’ATP qui constitue le site actif de l’enzyme (Lynch et coll., 2004). Elles entraînent une activation constitutive de la kinase, avec des différences d’effet en fonction de la nature et de la position de la mutation. D’autres modes d’activation oncogénique sont également décrits, tels que l’amplification génique ou la surexpression. Il semble que la mutation de l’EGFR soit un événement précoce dans la cancérogenèse chez les non-fumeurs. Les mécanismes de mutagenèse responsables de ces mutations ne sont pas connus, on sait cependant que les mutations de l’EGFR sont associées à l’altération de voies de sauvegarde et de maintien de l’intégrité génomique dont la voie p53 (Mounawar et coll., 2007).Mutations de KRAS
Les mutations des membres de la famille RAS (HRAS, KRAS2, NRAS) sont communes dans de nombreux types de cancers. Dans les cancers broncho-pulmonaires, 90 % de ces mutations affectent le gène KRAS. Elles sont presque systématiquement localisées au codon 12. Cette mutation est détectable dans 20 à 30 % des ADC et plus rarement dans les SCC.
Les protéines Ras jouent un rôle de relais et d’amplificateur des signaux intracellulaires déclenchés par l’activation des récepteurs tyrosine kinase tels que l’EGFR. Dans les cellules normales au repos, Ras est présente à la face cytoplasmique de la membrane plasmique sous une forme inactive, liée au GDP. Suite à la stimulation par un signal extracellulaire, Ras est recrutée au niveau du récepteur et interagit avec des facteurs d’échange des nucléotides guanidiques entraînant sa conversion en une forme active, liée au GTP. L’hydrolyse du GTP ramène l’activité au niveau de base et la répétition rapide de ce cycle permet la démultiplication intracellulaire du signal généré par l’activation du récepteur. La mutation au codon 12 bloque la protéine Kras2 en configuration active, entraînant la production d’un signal constitutif, indépendant de l’activation des récepteurs en amont. En accord avec cette observation, les mutations de KRAS interviennent généralement dans les tumeurs dépourvues de mutations de l’EGFR. En effet, les deux protéines agissent de façon séquentielle dans les mêmes cascades et les conséquences de ces mutations pourraient donc être au moins partiellement identiques.
Le type moléculaire des mutations au codon 12 de KRAS diffère en fonction de l’histoire tabagique du patient : les mutations G vers T dominent chez les fumeurs (comme les mutations de TP53 induites par le benzo(a)pyrene), alors que les mutations G vers A sont plus fréquentes chez les non-fumeurs (Le Calvez et coll., 2005). De plus, les mutations de KRAS semblent prédominer dans les ADC des fumeurs et des ex-fumeurs, à la différence des mutations de l’EGFR, que l’on trouve principalement chez les non-fumeurs. Cette observation suggère que les mêmes voies de signalisation pro-oncogéniques peuvent être activées de façon différente en fonction de l’étiologie et de l’histoire naturelle du cancer.
). De plus, les mutations de KRAS semblent prédominer dans les ADC des fumeurs et des ex-fumeurs, à la différence des mutations de l’EGFR, que l’on trouve principalement chez les non-fumeurs. Cette observation suggère que les mêmes voies de signalisation pro-oncogéniques peuvent être activées de façon différente en fonction de l’étiologie et de l’histoire naturelle du cancer.Altérations de la voie Rb
La protéine Rb, produit du gène du rétinoblastome RB1 (chromosome 13q14), est la clé de voûte d’une voie signalétique systématiquement altérée dans les cancers pulmonaires. Ce suppresseur de tumeurs agit comme facteur limitant pour contrôler la progression des cellules dans les phases G1 et S du cycle cellulaire. L’inactivation de ce « garde-barrière » est donc un exercice obligé pour mettre en place un processus de prolifération intempestive. Les mécanismes les plus communs sont la perte d’expression de RB1, l’extinction du gène INK4 (aussi décrit sous l’acronyme CDKN2a, chromosome 9p21) par méthylation de son promoteur, et la surexpression de la Cycline D1, produit du gène CCND1 (chromosome 11q13), souvent consécutive à l’amplification génique. Ces trois facteurs agissent de façon séquentielle dans la même cascade et régulent l’inhibition de Rb par phosphorylation. INK4 code pour p16, un inhibiteur des kinases cycline-dépendantes qui phosphorylent et inactivent Rb à la transition entre la phase G1 et la phase S du cycle cellulaire. La Cycline D1 est une des principales cyclines associées à ces kinases. La perte de l’expression de p16, l’amplification de CCND1 et l’inactivation de Rb ont donc essentiellement les mêmes conséquences et ne sont donc pas additives. Le locus INK4 est un site fréquent de perte d’allèles. De plus, dans les SCC des fumeurs, l’allèle résiduel est souvent hyper-méthylé, entraînant une inactivation fonctionnelle de l’expression de p16.
Inactivation de p14ARF
Le locus INK4/CDKN2a possède une structure complexe : il contient, en plus des séquences codant pour p16, un cadre de lecture pour une autre protéine, p14ARF, impliquée dans la répression de la prolifération par un mécanisme distinct de celui de p16. Dans la plupart des cas, la délétion du locus INK4 inactive à la fois p16 et p14ARF. Dans d’autres cancers, l’une ou l’autre protéine peut être inactivée de façon spécifique, suite à la méthylation différentielle des promoteurs qui gouvernent leur expression. La protéine p14ARF interagit avec Mdm2, le principal régulateur de la stabilité et de l’activité de p53. En se fixant à Mdm2, p14ARF stabilise p53 et induit une suppression de la prolifération cellulaire. Ce mécanisme fonctionne dans les cellules normales comme « garde-fou » contre la prolifération cellulaire intempestive ou excessive. Dès lors, p14ARF constitue donc une pièce centrale du carrefour de régulation décrit à la figure 3.3.
Voie Wnt/BetaCatenine/MYC
Les facteurs de la famille Wnt sont des protéines sécrétées impliquées dans la régulation de la prolifération, de la morphogenèse, de l’adhésion cellulaire, de la différenciation et de l’apoptose. Ils se lient à une famille de récepteurs de surface, Frizzled (Fzl), dont la stimulation induit une cascade signalétique impliquant la stabilisation de la beta-Caténine, sa translocation dans le noyau et l’activation des facteurs de transcription TCF, avec pour résultat l’augmentation de l’expression de nombreux gènes dont MYC et CCND1. Cette cascade est souvent activée dans de nombreux cancers, notamment dans les cancers du foie et du colon-rectum. Les composants de cette cascade sont aussi altérés dans la cancérogenèse pulmonaire, mais de façon très hétérogène. La mutation de la beta-Caténine est un événement plutôt rare, de même que la mutation de son régulateur APC, quel que soit le type histologique. En revanche, l’amplification de MYC (8q21-23) est détectée dans près de 10 % des SCLC, et est particulièrement fréquente dans les stades pré-invasifs (30 %). Cette amplification pourrait être associée au caractère hautement mitotique de ces lésions.
Pertes d’allèles en 3p
Une des altérations génétiques les plus communes dans les carcinomes broncho-pulmonaires, quel que soit leur type histologique, est la perte d’allèles dans la région p14-23 du chromosome 3, observée dans près de 80 % des NSCLC et des SCLC. Cette région chromosomique contient plusieurs candidats gènes-suppresseurs, dont FHIT, RASSF1 et SEMA3B. FHIT (Fragile Histidine Triad) est localisé dans une région chromosomique hautement fragile, propice à la formation de délétions sous l’effet direct des agents cancérogènes de la fumée du tabac. Le gène FHIT code une protéine possédant une activité ADP-hydroxylase, dont la fonction exacte est inconnue. Elle pourrait intervenir dans la régulation des niveaux de nucléotides intracellulaires et exercer des effets multiples, tant sur l’activation de nombreuses voies où des nucléotides sont impliqués que dans le contrôle de la synthèse d’ADN. La protéine codée par RASSF1 est un régulateur négatif de l’activité des membres de la famille RAS. L’allèle résiduel est souvent hyper-méthylé, avec pour effet la perte quasi-totale de l’expression du gène. L’impact exact sur la dérégulation de Ras reste à évaluer. SEMA3B code la Sémaphorine 3B, une protéine sécrétée impliquée dans la neurogenèse et la morphogenèse épithéliale. Ici aussi, les informations moléculaires sont trop fragmentaires pour comprendre la contribution exacte de ces altérations à la cancérogenèse bronchique. Il est possible que l’altération fréquente de cette région soit une simple conséquence de l’extrême fragilité du locus FHIT sous l’effet des agents cancérogènes du tabac, et constitue en quelque sorte une « signature » moléculaire de l’exposition tabagique.
Altérations de MEN1
Le gène MEN1, localisé sur le chromosome 11q13, code la Ménine, une protéine très particulière qui fonctionne comme un modulateur de facteurs de transcription mitogéniques tels que JunD ou AP1. La mutation de MEN1 est associée à une activité mitogénique élevée. La transmission héréditaire d’un allèle MEN1 muté est responsable de la néoplasie endocrine multiple de type 1, un syndrome autosomal dominant caractérisé par la formation de lésions néoplasiques de la glande parathyroïde, du tissu endocrine entéro-pancréatique, et de la glande pituitaire antérieure. Des mutations somatiques et des pertes d’allèles de MEN1 sont observées dans la majorité des carcinoïdes bronchiques atypiques, mais pas dans les tumeurs neuro-endocrines de haut grade. Il s’agit de la seule altération génétique connue à ce jour qui distingue les SCLC des NSCLC.
Biomarqueurs et impact clinique
Le cancer du poumon est la première cause de décès par cancer dans le monde, avec plus d’un million de décès par an pour un total de 1,2 millions de cancers diagnostiqués. Cette mortalité très élevée découle du caractère généralement tardif du diagnostic autant que de la relative inefficacité des traitements. En effet, les données du programme SEER1
(Surveillance, Epidemiology and End Results) pour la période 1996-2004 indiquent qu’environ 25 % des cancers du poumon et des bronches sont diagnostiqués à un stade régional (atteinte ganglionnaire sans métastases) et 50 % à un stade avancé (stade à distance, tumeurs métastasiques). Les résultats de la première étude de survie du réseau Francim donnent, respectivement chez l’homme et la femme, une survie relative à 5 ans standardisée sur l’âge, de 12 et 16 % (Bossard et coll., 2007). Si les données de survie du SEER selon le stade montrent que la survie relative à 5 ans pour les cancers localisés (tumeur sans extension ganglionnaire ni métastase) est d’environ 50 %, elle reste encore faible pour les stades plus avancés : stade régional (20,6 %) et stade à distance (2,8 %).
). Si les données de survie du SEER selon le stade montrent que la survie relative à 5 ans pour les cancers localisés (tumeur sans extension ganglionnaire ni métastase) est d’environ 50 %, elle reste encore faible pour les stades plus avancés : stade régional (20,6 %) et stade à distance (2,8 %).Les protocoles de traitement actuels sont basés sur la résection chirurgicale, accompagnée ou non d’une chimiothérapie adjuvante. L’essai clinique IALT (International Adjuvant Lung Therapy) a récemment démontré un bénéfice faible mais réel de la chimiothérapie impliquant les dérivés du platine. Les protocoles les plus communément appliqués font intervenir des combinaisons du cisplatine avec la gemcitabine, l’irinotecan, les taxanes, ou la vinorelbine (Filipits et coll., 2007).
).Malgré des progrès récents dans la connaissance des mécanismes moléculaires, il reste urgent d’identifier et de valider des biomarqueurs pour la détection précoce des cancers et pour la prédiction des réponses thérapeutiques. Un progrès important dans ce sens est la mise en évidence de l’impact des mutations de l’EGFR chez les non-fumeurs. En effet, ces cancers montrent généralement des réponses favorables à une nouvelle classe d’agents de thérapie ciblée, les inhibiteurs de tyrosine kinase tels que l’erlotinib ou le gefinitib (Giaccone et Rodriguez, 2005). Même si ces agents ne sont efficaces que pour une petite catégorie de patients, leurs effets démontrent l’intérêt de l’identification d’autres biomarqueurs de la réponse clinique permettant de mieux adapter les traitements aux caractéristiques de chaque cancer. En ce qui concerne la détection précoce, la mise en évidence de mutations spécifiques associées au étapes précoces de la cancérogenèse (et caractéristiques de différents mécanismes de mutagenèse) représente un espoir considérable (Hung et coll., 2005). Le principal problème est de mettre au point des stratégies pour la détection et l’évaluation de ces altérations dans des biopsies de très petite taille, dans des cellules exfoliées obtenues par lavages bronchiques, ou par l’étude des fragments d’ADN contenus dans les expectorations (Wang et coll., 2006).
). Même si ces agents ne sont efficaces que pour une petite catégorie de patients, leurs effets démontrent l’intérêt de l’identification d’autres biomarqueurs de la réponse clinique permettant de mieux adapter les traitements aux caractéristiques de chaque cancer. En ce qui concerne la détection précoce, la mise en évidence de mutations spécifiques associées au étapes précoces de la cancérogenèse (et caractéristiques de différents mécanismes de mutagenèse) représente un espoir considérable (Hung et coll., 2005). Le principal problème est de mettre au point des stratégies pour la détection et l’évaluation de ces altérations dans des biopsies de très petite taille, dans des cellules exfoliées obtenues par lavages bronchiques, ou par l’étude des fragments d’ADN contenus dans les expectorations (Wang et coll., 2006).Bibliographie
[1] bossard n, velten m, remontet l, belot a, maarouf n, et coll.. Survival of cancer patients in France: a population-based study from The Association of the French Cancer Registries (FRANCIM).
Eur J Cancer. 2007;
43:149160
[2] filipits m, haddad v, schmid k, huynh a, dunant a, et coll.. Multidrug resistance proteins do not predict benefit of adjuvant chemotherapy in patients with completely resected non-small cell lung cancer: International Adjuvant Lung Cancer Trial Biologic Program.
Clin Cancer Res. 2007;
13:38923898
[3] giaccone g,, rodriguez ja. EGFR inhibitors: what have we learned from the treatment of lung cancer?.
Nat Clin Pract Oncol. 2005;
2:554561
[4] hanahan d, weinberg ra. The hallmarks of cancer.
Cell. 2000;
100:5770
[5] hung rj, van der ho, tavtigian sv, brennan p, boffetta p, and hashibe m. Perspectives on the molecular epidemiology of aerodigestive tract cancers.
Mutat Res. 2005;
592:102118
[6] le calvez f, mukeria a, hunt jd, kelm o, hung rj, et coll.. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers.
Cancer Res. 2005;
65:50765083
[7] lynch tj, bell dw, sordella r, gurubhagavatula s, okimoto ra, et coll.. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib.
N Engl J Med. 2004;
350:21292139
[8] mounawar m, mukeria a, le calvez f, hung rj, renard h, et coll.. Patterns of EGFR, HER2, TP53, and KRAS mutations of p14arf expression in non-small cell lung cancers in relation to smoking history.
Cancer Res. 2007;
67:56675672
[9] pfeifer gd, denissenko mf, olivier m, tretyakova n, hecht ss, hainaut p. Tobacco smoke carcinogens, DNA-damage and p53 mutations in smoking-associated lung cancers.
Oncogene. 2002;
21:74357451
[10] shigematsu h, gazdar a. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers.
Int J Cancer. 2005;
118:257262
[11] wang yc, hsu hs, chen tp, chen jt. Molecular diagnostic markers for lung cancer in sputum and plasma.
Ann N Y Acad Sci. 2006;
1075:179184
[12] yokota j, kohno t. Molecular footprints of human lung cancer progression.
Cancer Sci. 2004;
95:197204
→ Aller vers le SOMMAIRE de l'ouvrage