Mécanismes généraux de toxicité
2008
| ANALYSE |
2-
Analyse de la toxicité de quelques polluants
Dans ce chapitre, nous passerons en revue quelques exemples d’agents chimiques et physiques qui ont fait l’objet d’études approfondies en toxicologie. Ces exemples illustrent les différents mécanismes d’action exposés précédemment.
Benzène
La toxicité du benzène sur le système hématopoïétique chez l’homme comme dans des modèles animaux est à présent reconnue. Ce polluant est aussi responsable de l’apparition de leucémies, en particulier des leucémies myéloïdes chroniques (LMC). Il est considéré comme cancérogène pour l’homme par le Centre international de recherche sur le cancer (Circ ou IARC en anglais, IARC, 1974 ). Malgré de nombreux travaux et des progrès récents, les mécanismes des effets leucémogènes chez l’homme demeurent controversés. En effet, selon un schéma classique pour des polluants organiques, la première hypothèse concernant cet hydrocarbure était que son métabolisme pourrait conduire à des composés réactifs susceptibles de réagir avec l’ADN, entraîner ainsi des mutations et l’apparition de clones cellulaires cancéreux. Cette hypothèse a été contestée principalement en raison de la faible génotoxicité du benzène et de ses métabolites dans des tests in vitro (Bird et coll., 2005). Pour analyser ce mécanisme, nous partirons de données acquises et tenterons de proposer un modèle cohérent.
). Malgré de nombreux travaux et des progrès récents, les mécanismes des effets leucémogènes chez l’homme demeurent controversés. En effet, selon un schéma classique pour des polluants organiques, la première hypothèse concernant cet hydrocarbure était que son métabolisme pourrait conduire à des composés réactifs susceptibles de réagir avec l’ADN, entraîner ainsi des mutations et l’apparition de clones cellulaires cancéreux. Cette hypothèse a été contestée principalement en raison de la faible génotoxicité du benzène et de ses métabolites dans des tests in vitro (Bird et coll., 2005). Pour analyser ce mécanisme, nous partirons de données acquises et tenterons de proposer un modèle cohérent.
). Malgré de nombreux travaux et des progrès récents, les mécanismes des effets leucémogènes chez l’homme demeurent controversés. En effet, selon un schéma classique pour des polluants organiques, la première hypothèse concernant cet hydrocarbure était que son métabolisme pourrait conduire à des composés réactifs susceptibles de réagir avec l’ADN, entraîner ainsi des mutations et l’apparition de clones cellulaires cancéreux. Cette hypothèse a été contestée principalement en raison de la faible génotoxicité du benzène et de ses métabolites dans des tests in vitro (Bird et coll., 2005). Pour analyser ce mécanisme, nous partirons de données acquises et tenterons de proposer un modèle cohérent.Cibles cellulaires du benzène
Plusieurs arguments montrent que les cellules progénitrices de la moëlle osseuse sont les cibles privilégiées du benzène et expliquent sa toxicité hématologique (Smith, 1996). En effet, la plupart des lignées sanguines sont touchées, ce qui suggère un effet en amont sur des cellules progénitrices. D’autre part, des études chez l’homme et chez l’animal montrent que le nombre de ces cellules et leurs fonctions sont diminuées et atteintes par le polluant. Enfin, même si les LMC sont les cancers hématologiques les plus souvent retrouvés, d’autres cancers sanguins ont été rapportés chez les personnes exposées.
). En effet, la plupart des lignées sanguines sont touchées, ce qui suggère un effet en amont sur des cellules progénitrices. D’autre part, des études chez l’homme et chez l’animal montrent que le nombre de ces cellules et leurs fonctions sont diminuées et atteintes par le polluant. Enfin, même si les LMC sont les cancers hématologiques les plus souvent retrouvés, d’autres cancers sanguins ont été rapportés chez les personnes exposées.Rôle du métabolisme
Le benzène est métabolisé d’abord par le cytochrome P4502E1 (ou CYP2E1) hépatique et sans doute d’autres enzymes hépatiques. Ces premières étapes conduisent à l’apparition d’oxyde de benzène, de 1,4 hydroquinone et de catéchol (Vaughan et coll., 2005). Ces composés peuvent subir d’autres étapes métaboliques dans le foie grâce notamment aux enzymes de phase 2 ce qui pourrait les détoxiquer. Ils peuvent sortir de l’hépatocyte périportal et atteindre la moëlle osseuse. Dans le compartiment myéloïde, la myélopéroxydase (MPO) transforme l’hydroquinone en benzoquinones. Ces composés ont des effets propres, mais peuvent aussi entrer dans un cycle d’oxydo-réduction conduisant à l’apparition d’un stress oxydant, sauf s’ils sont détoxiqués in situ par les NADPH-quinone-oxydoréductases. Il faut souligner que le benzène est un ligand du récepteur AhR, ce récepteur étant particulièrement abondant dans les cellules progénitrices et nécessaire chez la souris à l’expression de la toxicité du benzène (Yoon et coll., 2002). Par ailleurs, le système hématopoïétique contient des activités enzymatiques susceptibles d’inverser les effets des enzymes de phase 2 comme des sulfotransférases. L’importance du métabolisme du benzène dans sa toxicité a été soulignée par les effets toxiques de certains métabolites (voir ci-dessous), et par l’étude des polymorphismes génétiques de certaines enzymes impliquées dans le métabolisme du benzène (Lan et coll., 2004). En effet, des polymorphismes du gène NQO1 semblent accroître la toxicité et la cancérogénicité du benzène (études chez l’homme et dans des modèles animaux) (Iskander et Jaiswal, 2005). De plus, des polymorphismes inactivateurs de la MPO semblent jouer un rôle protecteur. Comme souvent, il est nécessaire de confirmer ces observations dans plusieurs études indépendantes avant de tirer des conclusions fermes, mais elles consolident l’implication du métabolisme dans la toxicité hématologique et la cancérogénicité du benzène.
). Ces composés peuvent subir d’autres étapes métaboliques dans le foie grâce notamment aux enzymes de phase 2 ce qui pourrait les détoxiquer. Ils peuvent sortir de l’hépatocyte périportal et atteindre la moëlle osseuse. Dans le compartiment myéloïde, la myélopéroxydase (MPO) transforme l’hydroquinone en benzoquinones. Ces composés ont des effets propres, mais peuvent aussi entrer dans un cycle d’oxydo-réduction conduisant à l’apparition d’un stress oxydant, sauf s’ils sont détoxiqués in situ par les NADPH-quinone-oxydoréductases. Il faut souligner que le benzène est un ligand du récepteur AhR, ce récepteur étant particulièrement abondant dans les cellules progénitrices et nécessaire chez la souris à l’expression de la toxicité du benzène (Yoon et coll., 2002). Par ailleurs, le système hématopoïétique contient des activités enzymatiques susceptibles d’inverser les effets des enzymes de phase 2 comme des sulfotransférases. L’importance du métabolisme du benzène dans sa toxicité a été soulignée par les effets toxiques de certains métabolites (voir ci-dessous), et par l’étude des polymorphismes génétiques de certaines enzymes impliquées dans le métabolisme du benzène (Lan et coll., 2004). En effet, des polymorphismes du gène NQO1 semblent accroître la toxicité et la cancérogénicité du benzène (études chez l’homme et dans des modèles animaux) (Iskander et Jaiswal, 2005). De plus, des polymorphismes inactivateurs de la MPO semblent jouer un rôle protecteur. Comme souvent, il est nécessaire de confirmer ces observations dans plusieurs études indépendantes avant de tirer des conclusions fermes, mais elles consolident l’implication du métabolisme dans la toxicité hématologique et la cancérogénicité du benzène.Effets génotoxiques
Cette première hypothèse a dû être rééxaminée étant donnée la faible mutagénicité du benzène et de ses métabolites principaux. Cependant, d’autres effets des métabolites du benzène sur l’ADN ont été rapportés, notamment leur capacité à entraîner des cassures de l’ADN. Cet effet est cohérent avec un certain nombre d’observations in vitro et in vivo. Ainsi, le test des comètes qui teste la présence de cassures est clairement positif. D’autre part, dans les leucémies liées à l’exposition au benzène, les translocations chromosomiques sont très fréquentes (Smith, 1996). Ces translocations touchent particulièrement certains chromosomes et pourraient correspondre à des sites de clivage privilégiés. Un travail récent s’est intéressé aux sites de coupure au niveau du gène MLL, qui est susceptible d’entraîner des réarrangements géniques. On observe parfois, mais moins souvent, des aneuploïdies et des mutations.
). Ces translocations touchent particulièrement certains chromosomes et pourraient correspondre à des sites de clivage privilégiés. Un travail récent s’est intéressé aux sites de coupure au niveau du gène MLL, qui est susceptible d’entraîner des réarrangements géniques. On observe parfois, mais moins souvent, des aneuploïdies et des mutations.Cibles protéiques
De nombreux travaux ont été consacrés aux cibles protéiques du benzène et de ses métabolites pouvant expliquer leurs effets sur l’intégrité de l’ADN. Les plus convaincants indiquent que la topoisomérase II pourrait être inhibée par certains métabolites, de manière analogue à certains agents alkylants eux-mêmes à l’origine de leucémies (Eastmond et coll., 2005). Cette enzyme modifie la topologie de l’ADN et, au cours de son cycle catalytique, elle entraîne une coupure d’un brin de l’ADN. Si des molécules interfèrent avec cette activité, elles pourraient stabiliser cette coupure et conduire aux effets génotoxiques observés.
). Cette enzyme modifie la topologie de l’ADN et, au cours de son cycle catalytique, elle entraîne une coupure d’un brin de l’ADN. Si des molécules interfèrent avec cette activité, elles pourraient stabiliser cette coupure et conduire aux effets génotoxiques observés.Stress oxydant
Le métabolisme du benzène est associé à l’apparition d’un stress oxydant qui pourrait avoir diverses origines : cycle d’oxydo-réduction des quinones/ hydroquinones, activation du récepteur AhR et induction de cytochromes P450... (Hirabayashi, 2005). Le stress oxydant pourrait être à l’origine des effets génotoxiques observés.
). Le stress oxydant pourrait être à l’origine des effets génotoxiques observés.Effets cellulaires
Des travaux récents indiquent des effets contradictoires sur l’apoptose et l’anoikis (détachement des cellules de leur substrat). Cependant, la possible inhibition de l’apoptose pourrait contribuer aux effets leucémogènes du benzène (Vaughan et coll., 2005). D’autres travaux ont montré un effet du benzène et/ou de ses métabolites sur les interactions cellulaires (Rivedal et Witz, 2005). Les effets cellulaires du benzène sont confortés par plusieurs expériences de génomique et de protéomique qui montrent une régulation des gènes impliqués dans le cycle cellulaire et l’apoptose (Hirabayashi, 2005; Smith et coll., 2005; Vermeulen et coll., 2005).
). D’autres travaux ont montré un effet du benzène et/ou de ses métabolites sur les interactions cellulaires (Rivedal et Witz, 2005). Les effets cellulaires du benzène sont confortés par plusieurs expériences de génomique et de protéomique qui montrent une régulation des gènes impliqués dans le cycle cellulaire et l’apoptose (Hirabayashi, 2005; Smith et coll., 2005; Vermeulen et coll., 2005).En résumé, l’ensemble de ces observations indique que les effets cancérogènes du benzène sont complexes et multiples. Nous pouvons proposer l’hypothèse suivante : les effets du benzène dépendent de son métabolisme hépatique puis de son métabolisme dans les cellules progénitrices de la moëlle osseuse. Le rôle activateur de la myélopéroxydase semble important puisqu’il conduit à l’apparition de quinone, la NQO1 étant plutôt protectrice dans la mesure où elle réduit ces quinones. Les métabolites sont susceptibles d’entraîner la coupure de l’ADN d’une part en inhibant la topoisomérase II et d’autre part en provoquant un stress oxydant. Certains métabolites inhibent l’apoptose et perturbent le cycle cellulaire, soit en raison de leurs effets sur l’ADN soit par des effets épigénétiques. Ces derniers pourraient être liés à l’activation du récepteur AhR ou au stress oxydant. Ce mécanisme complexe est cohérent avec la plupart des observations expérimentales, mais il semble encore difficile de hiérarchiser les différents événements. Dans l’état actuel des connaissances, il paraît peu probable que la toxicité du benzène soit associée à un seul métabolite et à un seul mécanisme, comme c’est le cas pour d’autres polluants.
Perturbateurs endocriniens : pesticides organochlorés et autres xénoœstrogènes
Les organochlorés sont des produits industriels aujourd’hui interdits ou en voie de l’être mais qui persistent encore dans l’environnement par leur stabilité et/ou leur emploi illégal. Ils comprennent les biphényles polychlorés (PCB, polychlorobiphényles), des pesticides (dichlorodiphényl-trichloroéthane ou DDT, lindane, chlordane, dieldrine, mirex), ainsi que les polychlorodibenzo-p-dioxines et polychlorodibenzofuranes (PCDD/F) qui sont des sous-produits générés lors de divers processus industriels ainsi que durant la combustion de déchets. Ces composés très stables sont solubles dans les graisses, ce qui fait qu’ils s’accumulent dans les tissus adipeux de différentes espèces. La bioamplification liée à la chaîne alimentaire fait que les humains qui consomment les espèces animales sont donc également exposés. Les concentrations augmentent avec l’âge et sont plus élevées dans les lipides de l’organisme, incluant les lipides sanguins, le tissu adipeux et les lipides du lait maternel. Certains organochlorés possèdent des propriétés œstrogéniques ou anti-androgènes tandis que d’autres montrent plutôt des effets anti-œstrogéniques (Ayotte et coll., 1994; Key et Reeves, 1994; Adami et coll., 1995; Safe et Zacharewski, 1997; Wolff et Weston, 1997; Laden et Hunter, 1998; Calle et coll., 2002; Mitra et coll., 2004; Kortenkamp, 2006). La notion d’effet systémique est renforcée par l’absence de concentration différentielle de ces produits dans le tissu mammaire cancéreux (Laden et Hunter, 1998).
; Key et Reeves, 1994; Adami et coll., 1995; Safe et Zacharewski, 1997; Wolff et Weston, 1997; Laden et Hunter, 1998; Calle et coll., 2002; Mitra et coll., 2004; Kortenkamp, 2006). La notion d’effet systémique est renforcée par l’absence de concentration différentielle de ces produits dans le tissu mammaire cancéreux (Laden et Hunter, 1998).Nous évoquerons ultérieurement le cas des dioxines. Dans cette section, nous discuterons des pesticides organochlorés et de leur relation avec les cancers hormono-dépendants. Les tests biologiques fondés sur des réponses utérines montrent que l’effet œstrogène de ces substances est corrélé à la constante d’affinité (Kd) pour le récepteur des œstrogènes (ER) (Jordan et coll., 1985; Safe et Zacharewski, 1997; Davidson, 1998; Jaga, 2000; Matthews et Zacharewski, 2000; Snedeker, 2001; Tapiero et coll., 2002; Starek, 2003; Bretveld et coll., 2006). Ce mode d’action est illustré dans la figure 2.1. Il en est de même pour les métaux œstrogéniques comme le cadmium, présent en milieu industriel et dans la fumée de cigarettes (Stoica et coll., 2000; Johnson et coll., 2003; Nesatyy et coll., 2006; Brama et coll., 2007). Le cadmium inhibe en outre l’expression du suppresseur de tumeur p53 (Meplan et coll., 1999). Il a été mis en cause dans les cancers du sein (Antila et coll., 1996; McElroy et coll., 2006) et de l’ovaire (Philipp et Hughes, 1982).
; Safe et Zacharewski, 1997; Davidson, 1998; Jaga, 2000; Matthews et Zacharewski, 2000; Snedeker, 2001; Tapiero et coll., 2002; Starek, 2003; Bretveld et coll., 2006). Ce mode d’action est illustré dans la figure 2.1. Il en est de même pour les métaux œstrogéniques comme le cadmium, présent en milieu industriel et dans la fumée de cigarettes (Stoica et coll., 2000; Johnson et coll., 2003; Nesatyy et coll., 2006; Brama et coll., 2007). Le cadmium inhibe en outre l’expression du suppresseur de tumeur p53 (Meplan et coll., 1999). Il a été mis en cause dans les cancers du sein (Antila et coll., 1996; McElroy et coll., 2006) et de l’ovaire (Philipp et Hughes, 1982).

Outre leurs effets cellulaires directs, les xéno-hormones pourraient aussi avoir un effet au niveau systémique conduisant à la perturbation du contrôle hormonal du développement de certains organes comme le testicule. Ces molécules peuvent inhiber la sécrétion des hormones hypophysaires par rétrocontrôle sur les récepteurs aux œstrogènes présents au niveau de l’hypothalamus (figure 2.2). L’action de ces xénoœstrogènes durant la période critique du développement fœtal correspondant à la morphogenèse testiculaire conduit au syndrome de dysgénésie testiculaire. En effet, il est admis que l’oligospermie, le cancer du testicule, la cryptorchidie et l’hypospadias soient les manifestations d’un même syndrome, le syndrome de dysgénésie testiculaire (SDT). Le SDT résulte de la perturbation hormonale du programme embryonnaire de développement des gonades durant la vie fœtale. Le SDT serait ainsi les conséquences d’une exposition anormale à des facteurs environnementaux, des perturbateurs endocriniens, dont les actions seraient probablement favorisées par un terrain génétique particulier.
). L’action de ces xénoœstrogènes durant la période critique du développement fœtal correspondant à la morphogenèse testiculaire conduit au syndrome de dysgénésie testiculaire. En effet, il est admis que l’oligospermie, le cancer du testicule, la cryptorchidie et l’hypospadias soient les manifestations d’un même syndrome, le syndrome de dysgénésie testiculaire (SDT). Le SDT résulte de la perturbation hormonale du programme embryonnaire de développement des gonades durant la vie fœtale. Le SDT serait ainsi les conséquences d’une exposition anormale à des facteurs environnementaux, des perturbateurs endocriniens, dont les actions seraient probablement favorisées par un terrain génétique particulier. | Figure 2.2 Inhibition de la sécrétion des hormones hypophysaires FSH et LH par les xénoœstrogènes (d’après Massaad et Barouki, 1999) |

Par l’intermédiaire d’un rétrocontrôle négatif, les xénoœstrogènes inhibent les sécrétions hypophysaires de FSH (Follicle Stimulating Hormone) et de LH (Luteinizing Hormone).
Il existe des composés naturels à effet œstrogénique. Les phytoœstrogènes alimentaires sont essentiellement présents dans les légumineuses (génistéine et daidzéine du soja) (Polkowski et Mazurek, 2000). Ils représentent 1 mg/kg de poids par jour chez l’adulte et 5-8 mg/kg chez les nourrissons suite à la consommation de formules lactées au soja (Setchell et coll., 1997). Les phytoœstrogènes sont agonistes des ERa et b in vitro et in vivo chez l’animal et chez l’homme (Benassayag et coll., 2002). La génistéine apparaît comme le chef de file des phytoœstrogènes, de par son affinité significative pour ERa (Kd = 100 nM), et sa proportion élevée (65 %) dans les phytoœstrogènes de soja. Cette affinité peut être préoccupante lorsqu’on sait que les taux plasmatiques de génistéine chez les enfants nourris au lait de soja sont 13 à 22 000 fois supérieurs à leur taux d’œstradiol circulant (Setchell et coll., 1998).
). Ils représentent 1 mg/kg de poids par jour chez l’adulte et 5-8 mg/kg chez les nourrissons suite à la consommation de formules lactées au soja (Setchell et coll., 1997). Les phytoœstrogènes sont agonistes des ERa et b in vitro et in vivo chez l’animal et chez l’homme (Benassayag et coll., 2002). La génistéine apparaît comme le chef de file des phytoœstrogènes, de par son affinité significative pour ERa (Kd = 100 nM), et sa proportion élevée (65 %) dans les phytoœstrogènes de soja. Cette affinité peut être préoccupante lorsqu’on sait que les taux plasmatiques de génistéine chez les enfants nourris au lait de soja sont 13 à 22 000 fois supérieurs à leur taux d’œstradiol circulant (Setchell et coll., 1998).Selon le travail pionnier de Lacassagne (Lacassagne, 1932), l’excès d’œstrogènes est capable d’induire le développement de tumeurs et pas seulement de promouvoir la croissance de cancers préexistants du fait d’un événement préalable (Eisinger et coll., 1999). Cet effet cancérogène pourrait aussi être secondaire à la perturbation du métabolisme hormonal provoqué par l’emploi de doses massives d’œstrogènes dans les expériences de Lacassagne et de ses contemporains. Il est généralement admis que la cancérogenèse œstrogéno-dépendante est liée à l’élévation des taux d’hormone circulante secondaire à un traitement pharmacologique ou toute autre cause jouant sur ce paramètre. Ainsi l’induction de l’aromatase ou l’inhibition des enzymes qui dégradent l’œstradiol peuvent conduire à des effets cancérogènes.
), l’excès d’œstrogènes est capable d’induire le développement de tumeurs et pas seulement de promouvoir la croissance de cancers préexistants du fait d’un événement préalable (Eisinger et coll., 1999). Cet effet cancérogène pourrait aussi être secondaire à la perturbation du métabolisme hormonal provoqué par l’emploi de doses massives d’œstrogènes dans les expériences de Lacassagne et de ses contemporains. Il est généralement admis que la cancérogenèse œstrogéno-dépendante est liée à l’élévation des taux d’hormone circulante secondaire à un traitement pharmacologique ou toute autre cause jouant sur ce paramètre. Ainsi l’induction de l’aromatase ou l’inhibition des enzymes qui dégradent l’œstradiol peuvent conduire à des effets cancérogènes.Toujours en relation avec l’idée de concentration excessive, certains organochlorés (dieldrine) sont des inducteurs enzymatiques et peuvent de ce fait modifier les voies de biotransformation de l’œstradiol, notamment la 16a-hydroxylation (Swaneck et Fishman, 1988). Cela conduit à la formation d’un métabolite se liant de façon covalente au récepteur des œstrogènes, qui pourrait prolonger l’action du complexe œstrogène-récepteur dans le noyau. Les xénoœstrogènes, cumulatifs ou non, sont donc aussi susceptibles de jouer un rôle prépondérant dans la régulation de la prolifération cellulaire du tissu mammaire.
). Cela conduit à la formation d’un métabolite se liant de façon covalente au récepteur des œstrogènes, qui pourrait prolonger l’action du complexe œstrogène-récepteur dans le noyau. Les xénoœstrogènes, cumulatifs ou non, sont donc aussi susceptibles de jouer un rôle prépondérant dans la régulation de la prolifération cellulaire du tissu mammaire.Hydrocarbures aromatiques polycycliques et/ou halogénés
En 1976, l’explosion de l’usine de pesticides ICMESA de Seveso (Italie) libéra dans l’atmosphère de grandes quantités de dioxines dont la dioxine prototype, dite « de Seveso » : 2,3,7,8-tétrachlorodibenzo-p-dioxine (TCDD). Ces dioxines furent générées par la pyrolyse de trichlorophénols. Deux cohortes ont été définies pour assurer la surveillance épidémiologique de cette population qui fut sévèrement exposée. Le groupe de Bertazzi mène des campagnes biennales depuis les années 1980 (Bertazzi et coll., 1989). Ils ont observé une modification du sex ratio en faveur des filles ainsi que des variations progressives de certains risques de pathologies (cancers, diabète) (Pesatori et coll., 2003). La cohorte du Seveso Women’s Health Study comporte 981 femmes résidantes des zones d’exposition les plus contaminées de Seveso et âgées de moins de 40 ans au moment de l’accident. Cette équipe a détecté un lien entre exposition aux dioxines et risque de cancer du sein (Eskenazi et coll., 2003). À la suite de cet accident, de nombreuses études ont exploré le rôle du récepteur des arylhydrocarbures (AhR), qui est activé non seulement par les arylhydrocarbures (benzo-a-pyrène, 3-méthylcholanthrène, diméthyl-7,12-benzanthracène) mais aussi par les hydrocarbures halogénés comme les dioxines et les PCB, dans une variété de mécanismes conduisant à la perturbation hormonale et aux cancers. Les auteurs des études épidémiologiques restent encore prudents sur l’existence du lien entre dioxines et cancers et son incidence sur les cancers humains du sein ou de l’ovaire (Kogevinas, 2001).
). Ils ont observé une modification du sex ratio en faveur des filles ainsi que des variations progressives de certains risques de pathologies (cancers, diabète) (Pesatori et coll., 2003). La cohorte du Seveso Women’s Health Study comporte 981 femmes résidantes des zones d’exposition les plus contaminées de Seveso et âgées de moins de 40 ans au moment de l’accident. Cette équipe a détecté un lien entre exposition aux dioxines et risque de cancer du sein (Eskenazi et coll., 2003). À la suite de cet accident, de nombreuses études ont exploré le rôle du récepteur des arylhydrocarbures (AhR), qui est activé non seulement par les arylhydrocarbures (benzo-a-pyrène, 3-méthylcholanthrène, diméthyl-7,12-benzanthracène) mais aussi par les hydrocarbures halogénés comme les dioxines et les PCB, dans une variété de mécanismes conduisant à la perturbation hormonale et aux cancers. Les auteurs des études épidémiologiques restent encore prudents sur l’existence du lien entre dioxines et cancers et son incidence sur les cancers humains du sein ou de l’ovaire (Kogevinas, 2001).Récepteur des arylhydrocarbures
Les arylhydrocarbures (Ah), les PCB coplanaires et les dioxines et furanes sont les principaux ligands du récepteur AhR. AhR reconnaît plus de 250 ligands dont 75 dioxines, mais le ligand naturel reste inconnu malgré quelques molécules candidates (Denison et Nagy, 2003). Les ligands AhR ont une affinité pour AhR qui varie entre 2-8 nM (dioxines) et 10-20 nM (arylhydrocarbures) selon les espèces (Collins et Marletta, 1984; Perdew et Hollenback, 1990). Un certain nombre de phytoœstrogènes sont des antagonistes du AhR tels la génistéine ou le resvératrol (Kd = 100 nM). Un modulateur physiologique antagoniste a été identifié, le 7-oxocholestérol (Savouret et coll., 2001). AhR est cytoplasmique en l’absence de ligand et il n’a aucune analogie avec les récepteurs stéroïdiens. Il se caractérise par un domaine N-terminal complexe bHLH-PAS1
, responsable de la liaison à l’ADN, de l’hétérodimérisation et de la liaison du ligand et des chaperonnes. Ce domaine définit une famille de transactivateurs qui comprend, outre AhR, son partenaire d’hétérodimérisation nucléaire ARNT (Aryl hydrocarbon Receptor Nuclear Translocator), le facteur de réponse à l’hypoxie HIF1a et le coactivateur des récepteurs stéroïdiens SRC-1 (Rowlands et Gustafsson, 1997). La partie C-terminale du récepteur AhR contient des régions transactivatrices et une fonction de répression de la transcription. AhR présente une rare aptitude aux interactions avec d’autres protéines telles que les chaperonnes (HSP90), le répresseur spécifique AhRR, le récepteur des œstrogènes, le corépresseur SMRT, des facteurs et cofacteurs de transcription et des tyrosines kinases (Carlson et Perdew, 2002). Toutes ces interactions sont susceptibles de générer des effets de perturbation hormonale puisqu’elles concernent des voies de signalisation hormonale.
). Les ligands AhR ont une affinité pour AhR qui varie entre 2-8 nM (dioxines) et 10-20 nM (arylhydrocarbures) selon les espèces (Collins et Marletta, 1984; Perdew et Hollenback, 1990). Un certain nombre de phytoœstrogènes sont des antagonistes du AhR tels la génistéine ou le resvératrol (Kd = 100 nM). Un modulateur physiologique antagoniste a été identifié, le 7-oxocholestérol (Savouret et coll., 2001). AhR est cytoplasmique en l’absence de ligand et il n’a aucune analogie avec les récepteurs stéroïdiens. Il se caractérise par un domaine N-terminal complexe bHLH-PAS1
, responsable de la liaison à l’ADN, de l’hétérodimérisation et de la liaison du ligand et des chaperonnes. Ce domaine définit une famille de transactivateurs qui comprend, outre AhR, son partenaire d’hétérodimérisation nucléaire ARNT (Aryl hydrocarbon Receptor Nuclear Translocator), le facteur de réponse à l’hypoxie HIF1a et le coactivateur des récepteurs stéroïdiens SRC-1 (Rowlands et Gustafsson, 1997). La partie C-terminale du récepteur AhR contient des régions transactivatrices et une fonction de répression de la transcription. AhR présente une rare aptitude aux interactions avec d’autres protéines telles que les chaperonnes (HSP90), le répresseur spécifique AhRR, le récepteur des œstrogènes, le corépresseur SMRT, des facteurs et cofacteurs de transcription et des tyrosines kinases (Carlson et Perdew, 2002). Toutes ces interactions sont susceptibles de générer des effets de perturbation hormonale puisqu’elles concernent des voies de signalisation hormonale.Après interaction avec son ligand, AhR se libère des chaperonnes et le complexe migre du cytoplasme vers le noyau (figure 2.3). AhR s’associe avec ARNT pour réguler l’expression de certains gènes en se liant à l’ADN au niveau de séquences définies, les « Dioxin (ou Xenobiotic) Responsive Elements » (DRE, XRE). Ces éléments sont présents dans la séquence des gènes cibles comme les cytochromes P-450 1A (CYP1A1, monooxygénases de phase I) ou les enzymes de la phase II de la détoxification (gluthathione-S-transférases, UDP glucuronyl transférase…).
). AhR s’associe avec ARNT pour réguler l’expression de certains gènes en se liant à l’ADN au niveau de séquences définies, les « Dioxin (ou Xenobiotic) Responsive Elements » (DRE, XRE). Ces éléments sont présents dans la séquence des gènes cibles comme les cytochromes P-450 1A (CYP1A1, monooxygénases de phase I) ou les enzymes de la phase II de la détoxification (gluthathione-S-transférases, UDP glucuronyl transférase…).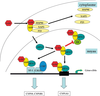 | Figure 2.3 Activation du récepteur AhR par ses ligands (dioxine, hydrocarbures aromatiques) conduisant à différents effets transcriptionnels ou interférentiels |
Le rôle des protéines induites par les dioxines dans la toxicité de ces dernières est sans doute très important. On a longtemps considéré que le récepteur AhR induisait principalement des enzymes du métabolisme des xénobiotiques dans le cadre de la réponse génique adaptative qui suit l’afflux de xénobiotiques potentiellement toxiques. Parmi ces dernières enzymes, les cytochromes P450 de la famille 1 sont les plus induits. Or, ces enzymes génèrent des ERO lors de leur cycle catalytique ce qui peut expliquer une partie de la toxicité de la dioxine (Barouki et Morel, 2001). Par ailleurs, ces enzymes métabolisent d’autres polluants comme des hydrocarbures aromatiques polycycliques (HAP) qui sont de bons activateurs du récepteur AhR. Or, certains métabolites des HAP sont particulièrement génotoxiques et ceci peut expliquer une part des effets cancérogènes des dioxines lorsqu’elles sont associées à des HAP.
). Par ailleurs, ces enzymes métabolisent d’autres polluants comme des hydrocarbures aromatiques polycycliques (HAP) qui sont de bons activateurs du récepteur AhR. Or, certains métabolites des HAP sont particulièrement génotoxiques et ceci peut expliquer une part des effets cancérogènes des dioxines lorsqu’elles sont associées à des HAP.Les travaux de toxicogénomique récents ont montré que les gènes des enzymes du métabolisme des xénobiotiques étaient loin d’être les seules cibles géniques du récepteur AhR (Frueh et coll., 2001). Plus de 150 gènes sont induits ou réprimés lorsque des cellules hépatiques humaines sont exposées à la dioxine. Il en est de même pour les macrophages humains ou le foie de souris (N’diaye et coll., 2006; Tijet et coll., 2006). Parmi les gènes induits, certains pourraient avoir un intérêt physiopathologique dans le cadre du cancer. Plusieurs cytokines sont induites et pourraient expliquer les effets inflammatoires associés à l’exposition aux dioxines ou aux HAP (Lecureur et coll., 2005). Plusieurs protéines impliquées dans le contrôle du cycle cellulaire sont aussi des cibles du récepteur AhR, mais il est difficile à ce stade de conclure quant aux effets finaux de ces régulations puisque certaines de ces protéines favorisent la croissance alors que d’autres auraient plutôt un effet antiprolifératif (Marchand et coll., 2005). Cette ambivalence dans le contrôle de la prolifération cellulaire est d’ailleurs assez fréquente lorsqu’on étudie les stress cellulaires qui ont souvent à la fois des effets proapoptotiques et des effets prolifératifs. Certains indiquent que ces effets sont sans doute liés puisque l’apoptose de cellules sensibles pourraient favoriser la prolifération d’autres cellules plus résistantes aux effets proapoptotiques. Enfin, parmi les gènes induits, on trouve des gènes impliqués dans la mobilité et les interactions cellulaires (Diry et coll., 2006). Cette observation est importante puisque l’invasion tumorale et l’apparition de métastases font appel à une mobilité et une plasticité cellulaires accrues. On peut faire l’hypothèse que les ligands du AhR pourraient favoriser la progression cancéreuse en plus des étapes initiales de la cancérogenèse. Cette proposition devrait être testée.
). Plus de 150 gènes sont induits ou réprimés lorsque des cellules hépatiques humaines sont exposées à la dioxine. Il en est de même pour les macrophages humains ou le foie de souris (N’diaye et coll., 2006; Tijet et coll., 2006). Parmi les gènes induits, certains pourraient avoir un intérêt physiopathologique dans le cadre du cancer. Plusieurs cytokines sont induites et pourraient expliquer les effets inflammatoires associés à l’exposition aux dioxines ou aux HAP (Lecureur et coll., 2005). Plusieurs protéines impliquées dans le contrôle du cycle cellulaire sont aussi des cibles du récepteur AhR, mais il est difficile à ce stade de conclure quant aux effets finaux de ces régulations puisque certaines de ces protéines favorisent la croissance alors que d’autres auraient plutôt un effet antiprolifératif (Marchand et coll., 2005). Cette ambivalence dans le contrôle de la prolifération cellulaire est d’ailleurs assez fréquente lorsqu’on étudie les stress cellulaires qui ont souvent à la fois des effets proapoptotiques et des effets prolifératifs. Certains indiquent que ces effets sont sans doute liés puisque l’apoptose de cellules sensibles pourraient favoriser la prolifération d’autres cellules plus résistantes aux effets proapoptotiques. Enfin, parmi les gènes induits, on trouve des gènes impliqués dans la mobilité et les interactions cellulaires (Diry et coll., 2006). Cette observation est importante puisque l’invasion tumorale et l’apparition de métastases font appel à une mobilité et une plasticité cellulaires accrues. On peut faire l’hypothèse que les ligands du AhR pourraient favoriser la progression cancéreuse en plus des étapes initiales de la cancérogenèse. Cette proposition devrait être testée.Outre les effets géniques du récepteur AhR, plusieurs groupes ont montré qu’il pouvait exercer ses effets en activant des cascades de signalisation différentes. Ainsi, plusieurs kinases semblent être la cible de ce récepteur, comme la p38 kinase et la JNK (Weiss et coll., 2005; Diry et coll., 2006). Dans le premier cas, l’activation est rapide et ne semble pas requérir l’induction de gènes alors que dans le deuxième, l’activation apparaît avec un délai compatible avec l’induction génique. La kinase Src est aussi activée par le récepteur AhR (Blankenship et Matsumura, 1997). Ces kinases sont impliquées dans des voies de stress cellulaire et il serait important d’évaluer leurs contributions aux effets cancérogènes du récepteur AhR.
; Diry et coll., 2006). Dans le premier cas, l’activation est rapide et ne semble pas requérir l’induction de gènes alors que dans le deuxième, l’activation apparaît avec un délai compatible avec l’induction génique. La kinase Src est aussi activée par le récepteur AhR (Blankenship et Matsumura, 1997). Ces kinases sont impliquées dans des voies de stress cellulaire et il serait important d’évaluer leurs contributions aux effets cancérogènes du récepteur AhR.Parmi les observations récentes sur le récepteur AhR, nous retiendrons la plasticité moléculaire de ce récepteur vis-à-vis de différents ligands dans la mesure où ces travaux pourraient conduire à des applications pharmacologiques. Plusieurs laboratoires ont observé que différents ligands de ce récepteur pouvaient conduire à des effets géniques distincts : par exemple, la dioxine, les HAP et les polyphénols, tous ligands de ce récepteur, ont des effets distincts en terme d’induction génique. Les polyphénols par exemple se comportent comme des antagonistes vis-à-vis de l’induction de certains gènes par la dioxine alors qu’ils ont des effets de type agonistes sur d’autres gènes (Casper et coll., 1999; Gouédard et coll., 2004). Safe a proposé le concept de SahRM pour Selective Ah Receptor Modulators (Safe et McDougal, 2002). Ces molécules pourraient avoir un intérêt pharmacologique dans la mesure où elles s’opposeraient aux effets toxiques des HAP et de la dioxine mais pourraient garder certaines propriétés protectrices comme l’activité anti-œstrogénique que nous n’avons pas discutée ici.
; Gouédard et coll., 2004). Safe a proposé le concept de SahRM pour Selective Ah Receptor Modulators (Safe et McDougal, 2002). Ces molécules pourraient avoir un intérêt pharmacologique dans la mesure où elles s’opposeraient aux effets toxiques des HAP et de la dioxine mais pourraient garder certaines propriétés protectrices comme l’activité anti-œstrogénique que nous n’avons pas discutée ici.Interactions dioxines-œstrogènes
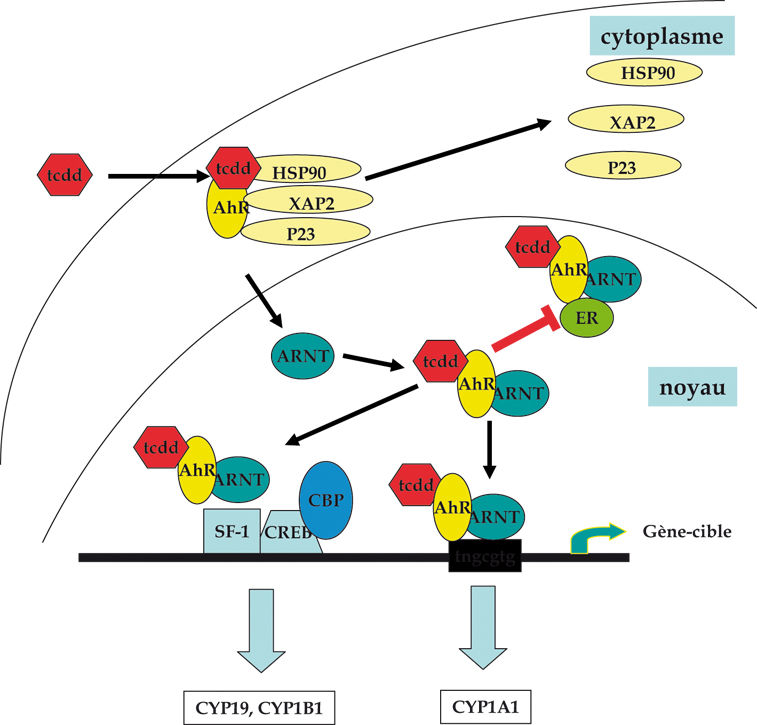
Étant donné l’implication des œstrogènes dans de nombreux cancers, notamment le cancer du sein, de nombreux travaux ont porté sur l’interaction entre dioxines (ou HAP) et voie de signalisation des œstrogènes.
Le couple AhR/dioxine présente des effets fortement anti-œstrogèniques sur différents gènes-cibles par interaction directe avec ER (Wormke et coll., 2003), ce qui rend d’ailleurs difficile à interpréter les récents résultats sur l’augmentation de fréquence du cancer du sein dans la cohorte Seveso d’Eskenazi (Eskenazi et coll., 2003). De plus, ces effets anti-œstrogéniques pourraient être indirects. Par exemple, AhR interagit avec le corépresseur SMRT, ce qui libère l’activité transactivatrice du récepteur rétinoique (Widerak et coll., 2005). Ce récepteur est considéré comme s’opposant aux effets prolifératifs du ER dans les cancers du sein (Darro et coll., 1998; Afonja et coll., 2002). Cependant, des travaux récents indiquent que les relations entre AhR et ER étaient assez complexes. Ainsi le groupe de Kato a montré dans un premier temps que le AhR activé pouvait interagir avec le récepteur ER en l’absence d’hormones et induire ainsi des gènes œstrogéno-dépendants (Ohtake et coll, 2003). Dans un deuxième temps, ce groupe a montré que le récepteur AhR exerçait à l’égard du ER une activité « ubiquitine E3 ligase » et orientait le ER vers le protéasome favorisant ainsi sa dégradation (Ohtake et coll, 2007). Il résulte de ces derniers travaux que les dioxines perturbent considérablement les régulations hormonales œstrogéniques : en présence d’hormones, ces polluants exercent un rôle antagoniste, alors qu’en absence d’hormone, ils sont capables d’induire transitoirement des gènes hormono-dépendants (figure 2.4).
), ce qui rend d’ailleurs difficile à interpréter les récents résultats sur l’augmentation de fréquence du cancer du sein dans la cohorte Seveso d’Eskenazi (Eskenazi et coll., 2003). De plus, ces effets anti-œstrogéniques pourraient être indirects. Par exemple, AhR interagit avec le corépresseur SMRT, ce qui libère l’activité transactivatrice du récepteur rétinoique (Widerak et coll., 2005). Ce récepteur est considéré comme s’opposant aux effets prolifératifs du ER dans les cancers du sein (Darro et coll., 1998; Afonja et coll., 2002). Cependant, des travaux récents indiquent que les relations entre AhR et ER étaient assez complexes. Ainsi le groupe de Kato a montré dans un premier temps que le AhR activé pouvait interagir avec le récepteur ER en l’absence d’hormones et induire ainsi des gènes œstrogéno-dépendants (Ohtake et coll, 2003). Dans un deuxième temps, ce groupe a montré que le récepteur AhR exerçait à l’égard du ER une activité « ubiquitine E3 ligase » et orientait le ER vers le protéasome favorisant ainsi sa dégradation (Ohtake et coll, 2007). Il résulte de ces derniers travaux que les dioxines perturbent considérablement les régulations hormonales œstrogéniques : en présence d’hormones, ces polluants exercent un rôle antagoniste, alors qu’en absence d’hormone, ils sont capables d’induire transitoirement des gènes hormono-dépendants (figure 2.4). | Figure 2.4 Activation illégitime de la voie de signalisation du RE (récepteur des œstrogènes) par le récepteur de la dioxine activé (d’après Massaad et Barouki, 1999) |
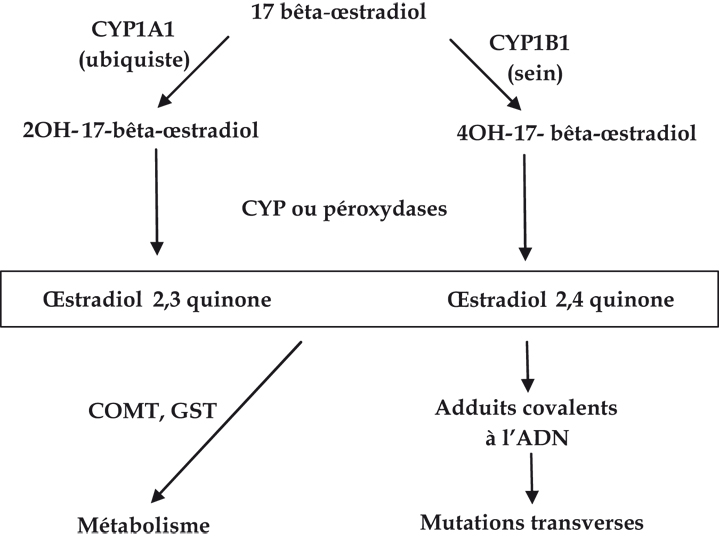
Outre les interactions entre les récepteurs AhR et ER, les dioxines peuvent modifier le métabolisme des œstrogènes et l’orienter dans un sens toxique. Différentes voies métaboliques peuvent conduire à la synthèse de composés génotoxiques à partir d’œstrogènes naturels. La voie principale (figure 2.5) conduit à la synthèse de dérivés hydroxylés de l’E2, les catéchols (Coumoul et Barouki, 2002). Il s’agit du 2CE (ou 2OH-E2 pour 2OH-catechol estrogen) qui est le principal catéchol dans le sang et l’urine, et du 4CE (ou 4OH-E2 pour 4OH-catechol estrogen) produit de façon majoritaire dans le sein, l’endomètre et l’ovaire (tumoral ou non). Ces composés sont synthétisés majoritairement par des cytochromes P450 (CYP) mais aussi par d’autres enzymes comme l’aromatase ou certaines péroxydases. Les différences de production des catéchols selon les tissus sont expliquées par le fait que l’expression des différents cytochromes P450 est variable selon les tissus. Ainsi, l’œstradiol est converti en 2CE par CYP1A et en 4CE par CYP1B1 (non-hépatique). Les catéchols conduisent à la synthèse de semi-quinones puis à celle des quinones (CYP ou péroxydases puis mécanismes de super oxydation non-enzymatique). La voie principale de détoxication des catéchols fait intervenir la catéchol O-méthyl transférase (COMT) ou les glutathion-S-transférases (GST).
) conduit à la synthèse de dérivés hydroxylés de l’E2, les catéchols (Coumoul et Barouki, 2002). Il s’agit du 2CE (ou 2OH-E2 pour 2OH-catechol estrogen) qui est le principal catéchol dans le sang et l’urine, et du 4CE (ou 4OH-E2 pour 4OH-catechol estrogen) produit de façon majoritaire dans le sein, l’endomètre et l’ovaire (tumoral ou non). Ces composés sont synthétisés majoritairement par des cytochromes P450 (CYP) mais aussi par d’autres enzymes comme l’aromatase ou certaines péroxydases. Les différences de production des catéchols selon les tissus sont expliquées par le fait que l’expression des différents cytochromes P450 est variable selon les tissus. Ainsi, l’œstradiol est converti en 2CE par CYP1A et en 4CE par CYP1B1 (non-hépatique). Les catéchols conduisent à la synthèse de semi-quinones puis à celle des quinones (CYP ou péroxydases puis mécanismes de super oxydation non-enzymatique). La voie principale de détoxication des catéchols fait intervenir la catéchol O-méthyl transférase (COMT) ou les glutathion-S-transférases (GST).La production des catéchols dépend des activités des enzymes qui les produisent, qui elles-mêmes dépendent du tissu étudié et de la présence ou non d’inducteurs. Lorsque la synthèse de catéchols devient excessive, les systèmes de détoxication (COMT, sulfotransférases, UDP-glucuronosyltransférases) sont dépassés et les dérivés, semi-quinones et quinones, sont produits. La formation des quinones à partir des semi-quinones peut conduire à la formation d’espèces réactives de l’oxygène (ERO). La production excessive de ERO endommage l’ADN (Park et coll., 1996) mais aussi les lipides et les protéines. Le 2CE est moins toxique que le 4CE, voire protecteur. Seul le 4CE est expérimentalement cancérogène par formation d’adduits covalents sur les bases puriques de l’ADN (4OH-E2-1-N7-guanine et 4OH-E2-1-N3-adénosine). Les adduits formés par le 2CE sont stables et peu mutagènes, contrairement à ceux provoqués par le 4CE. En outre, une activité 4_hydroxylase élevée, liée à CYP1B1, est détectée dans le tissu mammaire ou ovarien tumoral (Liehr et coll., 1986; Liehr, 1997 et 2001).
) mais aussi les lipides et les protéines. Le 2CE est moins toxique que le 4CE, voire protecteur. Seul le 4CE est expérimentalement cancérogène par formation d’adduits covalents sur les bases puriques de l’ADN (4OH-E2-1-N7-guanine et 4OH-E2-1-N3-adénosine). Les adduits formés par le 2CE sont stables et peu mutagènes, contrairement à ceux provoqués par le 4CE. En outre, une activité 4_hydroxylase élevée, liée à CYP1B1, est détectée dans le tissu mammaire ou ovarien tumoral (Liehr et coll., 1986; Liehr, 1997 et 2001). | Figure 2.5 Métabolisme de l’œstradiol par les cytochromes CYP1A/1B conduisant à la mutagenèse procarcinogène dans le cas de la quinone 2,4 de l’œstradiol |
Perturbateurs métaboliques : les phtalates
Les phtalates conduisent à des tumeurs hépatocellulaires dans les modèles animaux, et à des tumeurs du testicule après exposition en continu. Aucun effet génotoxique direct n’a été démontré in vitro. Les mécanismes proposés sont des effets œstrogéniques ou l’activation de PPARa (Peroxisome Proliferator-Activated Receptor alpha). Selon cette dernière hypothèse, l’activation de PPARa conduirait à la fois à un stress oxydatif qui causerait des dommages de l’ADN de type cassures simple-brin potentiellement mutagènes si elles ne sont pas réparées, et à l’expression de gènes impliqués dans la prolifération cellulaire. Ces actions combinées pourraient favoriser le développement de foyers pré-néoplasiques puis des tumeurs (Rusyn et coll., 2006). Cette hypothèse est cependant contestée. Par ailleurs, il a été récemment montré que les phtalates activaient aussi le récepteur PPARg notamment au niveau adipocytaire (Feige et coll., 2007). Ces composés sont donc des perturbateurs métaboliques globaux qui pourraient avoir des effets sur la croissance et la survie des cellules.
). Cette hypothèse est cependant contestée. Par ailleurs, il a été récemment montré que les phtalates activaient aussi le récepteur PPARg notamment au niveau adipocytaire (Feige et coll., 2007). Ces composés sont donc des perturbateurs métaboliques globaux qui pourraient avoir des effets sur la croissance et la survie des cellules.Métaux
Les mécanismes d’action du cadmium et de l’arsenic sont envisagés.
Cadmium
Le cadmium (Cd) est un métal lourd reconnu comme cancérogène par le Circ (Centre international de recherche sur le cancer). Il est retrouvé sous forme d’oxydes dans les batteries et catalyseurs, de sulfides dans les pigments, de sulfates utilisés dans la métallisation et de stéarates utilisés comme stabilisants de plastiques.
L’exposition est essentiellement sous forme de poussières et de fumée. Les fortes expositions sont d’origine professionnelle (industries de production, métallisation). Les autres sources pour la population non exposée sont la fumée de cigarette et les aliments (riz) dans certaines zones contaminées.
L’entrée dans l’organisme se fait par ingestion ou par inhalation. La fraction absorbée au niveau intestinal est influencée par des facteurs diététiques. La fraction absorbée au niveau pulmonaire dépend de la solubilité du composé. Une fois dans l’organisme, le cadmium se retrouve lié à la métallothionéine. Il est essentiellement concentré au niveau hépatique et rénal.
Des modèles expérimentaux chez le rat ont permis d’étudier la cancérogénicité du cadmium. En particulier, l’administration de chlorure de cadmium (CdCl2) avec contrôle du taux de zinc, a conduit à une augmentation dose-dépendante de l’incidence de leucémie, de tumeurs à cellules interstitielles testiculaires et de lésions prolifératives de la prostate. En revanche, l’administration de CdCl2 sans contrôle du taux de zinc n’a pas conduit à une augmentation de l’incidence de cancer.
Le cadmium démontre une faible liaison à l’ADN. Les lésions de l’ADN ne sont donc pas le mode d’action principal. Le stress oxydatif pourrait constituer un mécanisme d’action bien que le Cd ne soit pas un métal redox actif. Par ce biais, il pourrait conduire à des dommages de l’ADN, mais ce mécanisme n’a pas encore été clairement démontré.
Des dommages de l’ADN ont été observés en culture cellulaire dans le cas de sulfides de Cd (aberrations chromosomiques), chlorures de Cd (aneuploïdie).
Le Cd étant peu mutagène, des mécanismes indirects ou épigénétiques ont été avancés, tels l’activation d’oncogènes/inhibition de l’apoptose, ou la diminution de la réparation de l’ADN endommagé.
Les poumons sont la cible la plus décrite et la plus certaine. Quelques études ont montré un lien avec le cancer du rein. La transformation de cellules épithéliales prostatiques suite à une exposition au Cd a été rapportée. Enfin, de fortes doses de Cd pourraient conduire à des tumeurs à cellules interstitielles testiculaires.
Arsenic
L’arsenic (As), chimiquement très proche du phosphore, est un cancérogène associé au cancer de la peau, des poumons, du foie, du rein et de la vessie (National Toxicology Program, 2000). Une association a également été décrite avec le cancer de la prostate suite à des expositions chroniques à de l’arsenic inorganique (Chen et Wang, 1990; Lewis et coll., 1999). Il a été démontré que l’arsenic peut conduire à une transformation de cellules épithéliales prostatiques humaines in vitro, et que ces cellules transformées conduisent à des tumeurs très agressives quand elles sont inoculées à la souris nude (Achanzar et coll., 2002).
). Une association a également été décrite avec le cancer de la prostate suite à des expositions chroniques à de l’arsenic inorganique (Chen et Wang, 1990; Lewis et coll., 1999). Il a été démontré que l’arsenic peut conduire à une transformation de cellules épithéliales prostatiques humaines in vitro, et que ces cellules transformées conduisent à des tumeurs très agressives quand elles sont inoculées à la souris nude (Achanzar et coll., 2002).La toxicité de l’arsenic dépend de son degré d’oxydation et de sa composition chimique. Alors que l’entrée de l’arsénite dans la cellule se fait par diffusion passive, l’influx de l’arséniate se fait par compétition avec le phosphate. L’arsénite est très réactif vis-à-vis des groupements thiols, et peut en se liant à des résidus cystéine proches du site actif de certaines enzymes comme les tyrosine phosphatases, les enzymes intervenant dans le processus d’ubiquitination, réduire leurs activités. L’arséniate de structure très proche du phosphate va plutôt interférer avec les réactions de phosphorylation oxydative en formant un ester d’arséniate instable. L’arséniate inhibe donc les réactions de transfert de groupement phosphate nécessaires à la régénération de l’ATP (Huang et coll., 2004). Par ailleurs, l’excrétion de l’arséniate est plus rapide que celle de l’arsénite. Ce dernier est par conséquent beaucoup plus toxique et cancérogène que l’arséniate.
). Par ailleurs, l’excrétion de l’arséniate est plus rapide que celle de l’arsénite. Ce dernier est par conséquent beaucoup plus toxique et cancérogène que l’arséniate.Une des conséquences de l’exposition à l’arsenic est la génération d’espèces réactives de l’oxygène (ERO). Ces ERO sont impliquées dans la cancérogenèse à la fois au niveau de l’étape d’initiation, par les dommages causés à l’ADN de type cassures simple-brin, et à l’étape de promotion, par les modifications des voies de signalisation intracellulaire comme celle de AP-1 et de NF-kB.
Fibres d’amiante
De nombreux échantillons de fibres d’amiante (amphiboles, chrysotile) ont été testés chez l’animal, par inhalation ou par injection intra-cavitaire (intra-pleurale ou intra-péritonéale), essentiellement chez le rat. Les résultats ont démontré un potentiel cancérogène des échantillons, lesquels provoquaient des tumeurs pulmonaires et des mésothéliomes ; ils ont permis de mettre en évidence des caractéristiques des fibres modulant la cancérogénicité. Par ailleurs, l’étude des effets sur différents systèmes de cellules en culture a permis de préciser certaines hypothèses sur le mécanisme d’action de ces fibres.
La toxicité des fibres d’amiante semble le fait de deux mécanismes ; l’un est associé à la réaction inflammatoire qui accompagne le dépôt des fibres dans les voies aériennes et le poumon. Il en résulte un afflux de cellules inflammatoires qui produisent des facteurs : espèces réactives dérivées de l’oxygène (ERO) ou de l’azote (ERN) et cytokines, et un contexte dans lequel les cellules doivent s’adapter à ce nouvel environnement (stress oxydant). Ces molécules sont en outre capables de produire des lésions de l’ADN, de modifier les protéines cellulaires et de favoriser la prolifération cellulaire. Une oxydation de bases (en particulier 8-hydroxy-déoxyguanosine, 8-OHdG) et des cassures d’ADN ont été détectées dans des cellules exposées à l’amiante, dont l’origine pourrait s’expliquer par ce mécanisme (Kane, 1999; Upadhyay et coll., 2003). La survenue de lésions de l’ADN a été aussi suggérée de manière indirecte, par la mise en évidence de l’activation de mécanismes de réparation de l’ADN ou par un arrêt du cycle cellulaire dans les cellules exposées à l’amiante (Jaurand, 1997 et 1999). De plus, certains facteurs produits au cours de la réaction inflammatoire sont susceptibles de provoquer une stimulation de la prolifération cellulaire. Cela a été mis en évidence in vivo, lors de l’exposition d’animaux aux fibres d’amiante et, sous certaines conditions, avec des cellules en culture (Mc Gavran et Brody, 1989; Adamson et coll., 1993; Dixon et coll., 1995; Driscoll, 1999). On conçoit que la prolifération de cellules épithéliales dont l’ADN présente des lésions non ou mal réparées aura pour conséquence un risque accru de transformation.
; Upadhyay et coll., 2003). La survenue de lésions de l’ADN a été aussi suggérée de manière indirecte, par la mise en évidence de l’activation de mécanismes de réparation de l’ADN ou par un arrêt du cycle cellulaire dans les cellules exposées à l’amiante (Jaurand, 1997 et 1999). De plus, certains facteurs produits au cours de la réaction inflammatoire sont susceptibles de provoquer une stimulation de la prolifération cellulaire. Cela a été mis en évidence in vivo, lors de l’exposition d’animaux aux fibres d’amiante et, sous certaines conditions, avec des cellules en culture (Mc Gavran et Brody, 1989; Adamson et coll., 1993; Dixon et coll., 1995; Driscoll, 1999). On conçoit que la prolifération de cellules épithéliales dont l’ADN présente des lésions non ou mal réparées aura pour conséquence un risque accru de transformation.Le second mécanisme de toxicité des fibres d’amiante, non exclusif du précédent, résulte de la capacité des cellules épithéliales, et des cellules mésothéliales, à internaliser les fibres d’amiante. Il a été montré que la phagocytose des fibres d’amiante était, elle aussi, associée à la génération d’ERO et d’ERN, et que la division cellulaire des cellules exposées à l’amiante était considérablement altérée (Jaurand, 1999; Jaurand et Levy, 1999; Kane, 1999; Wu et coll., 2000). Dans ce contexte, les cellules doivent réagir aux oxydants et s’adapter à la charge en particules qu’elles hébergent. Des perturbations de la mitose et des altérations chromosomiques ont été mises en évidence dans de nombreuses études, sur différents types cellulaires, incluant des cellules mésothéliales pleurales. Diverses altérations ont été mises en évidence : formation de cassures de chromosomes, anomalies de ségrégation des chromosomes, perte d’hétérozygotie (Lechner et coll., 1985; Wang et coll., 1987; Hei et coll., 1992; Both et coll., 1994 et 1995; Yegles et coll., 1993 et 1995; Dopp et coll., 1995 et 1997; Jensen et coll., 1996; Dopp et Schiffmann, 1998; Jensen et Watson, 1999; Poser et coll., 2004). Celles-ci ne résultent pas nécessairement d’un effet mécanique, mais peuvent résulter des lésions de l’ADN. On peut prévoir que ces effets auront des conséquences importantes sur le capital génétique des cellules, en termes de dosage et d’expression géniques (délétions, translocations, expression dérégulée…).
; Jaurand et Levy, 1999; Kane, 1999; Wu et coll., 2000). Dans ce contexte, les cellules doivent réagir aux oxydants et s’adapter à la charge en particules qu’elles hébergent. Des perturbations de la mitose et des altérations chromosomiques ont été mises en évidence dans de nombreuses études, sur différents types cellulaires, incluant des cellules mésothéliales pleurales. Diverses altérations ont été mises en évidence : formation de cassures de chromosomes, anomalies de ségrégation des chromosomes, perte d’hétérozygotie (Lechner et coll., 1985; Wang et coll., 1987; Hei et coll., 1992; Both et coll., 1994 et 1995; Yegles et coll., 1993 et 1995; Dopp et coll., 1995 et 1997; Jensen et coll., 1996; Dopp et Schiffmann, 1998; Jensen et Watson, 1999; Poser et coll., 2004). Celles-ci ne résultent pas nécessairement d’un effet mécanique, mais peuvent résulter des lésions de l’ADN. On peut prévoir que ces effets auront des conséquences importantes sur le capital génétique des cellules, en termes de dosage et d’expression géniques (délétions, translocations, expression dérégulée…).On s’est interrogé pour savoir quelles sont les caractéristiques des fibres qui modulent la réponse cellulaire. Il est clair que le potentiel cancérogène des fibres dépend de leurs dimensions et est lié à la forme de ces particules, mais ces seules caractéristiques des fibres ne semblent pas être les paramètres uniques responsables de la toxicité. Le rôle des dimensions a été mis en évidence in vivo, dans des études par inhalation ou par inoculation intra-cavitaire, ainsi que dans des études sur cellules en culture. Dans ces études, lorsque la comparaison des effets a été effectuée avec des échantillons de différentes dimensions, il a été généralement observé que les fibres longues étaient plus actives que les fibres courtes.
Concernant les dimensions, il est à noter que les fibres d’amiante présentent une spécificité de dépôt dans le poumon, par rapport à des particules granulaires. En effet, si pour des raisons physiques et dynamiques des particules granulaires de diamètre aérodynamique moyen supérieur à 5 mm ne sont pas déposées dans le poumon profond, des fibres de longueur supérieure à 5 mm peuvent atteindre les alvéoles pulmonaires, en raison de leur forme. Par ailleurs, il a été démontré que les fibres étaient susceptibles d’être transloquées vers la plèvre, ayant ainsi la possibilité d’interagir, non seulement avec les cellules épithéliales bronchiques et pulmonaires, mais également avec les cellules mésothéliales.
Les propriétés de surface des fibres sont un autre paramètre influençant leur réactivité. Un très grand nombre de travaux a porté sur les propriétés oxydo-réductrices ; elles sont associées à la présence de métaux, en particulier le fer, jouant un rôle de catalyseur, et susceptible de générer des ERO (s’ajoutant aux ERO générées par les différents types cellulaires (Shukla et coll., 2003).
).Outre la production d’ERO, les propriétés de surface des fibres leur confèrent la capacité à adsorber des macromolécules biologiques, protéines et ADN. L’adsorption de protéines, telles la vitronectine ou des protéines sériques est susceptible de modifier leur réactivité sur cultures cellulaires (phagocytose, production d’ERO). Des molécules organiques (hydrocarbures aromatiques polycycliques, HAP) ont également été détectées à la surface des fibres. L’explication de l’effet multiplicatif du tabac chez les sujets fumeurs exposés à l’amiante repose en partie sur l’hypothèse du potentiel des fibres à interagir avec les HAP. Il a été observé, chez le rat, qu’un traitement de surface de fibres de chrysotile (phosphatation) modifiait leur potentiel cancérogène (Van der Meeren et coll., 1992).
).La composition chimique des fibres d’amiante intervient également pour rendre compte de leur pouvoir cancérogène. Dans une étude portant sur la tumorigénicité de divers échantillons de chrysotile, il a été observé qu’une modification préalable de la composition chimique des fibres (solubilisation du magnésium par traitement acide) s’accompagnait d’une diminution de la cancérogénicité (Monchaux et coll., 1981).
).En résumé, le mécanisme d’action des fibres d’amiante est complexe. L’ensemble des données montre un potentiel génotoxique mis en évidence par des lésions de l’ADN et des chromosomes. Le potentiel des fibres à provoquer une transformation néoplasique des cellules dépendra, outre de la dose, des caractéristiques physiques et physico-chimiques des fibres.
Rayonnements ionisants
Jusqu’à ces dernières années, le dogme central de la radiobiologie classique reposait sur un modèle biologique simple. L’ADN était considéré comme la cible essentielle des rayonnements ionisants. Un dépôt d’énergie au niveau du noyau de la cellule peut entraîner des lésions au niveau de l’ADN, qui si elles sont incomplètement ou imparfaitement réparées, peuvent se traduire par une modification du patrimoine génétique de la cellule. Ces altérations constituent la première étape vers la transformation cancéreuse lorsqu’elles surviennent dans des cellules somatiques (figure 2.6).
). | Figure 2.6 Dogme classique des effets biologiques d’une exposition aux rayonnements ionisants au niveau de la cellule |
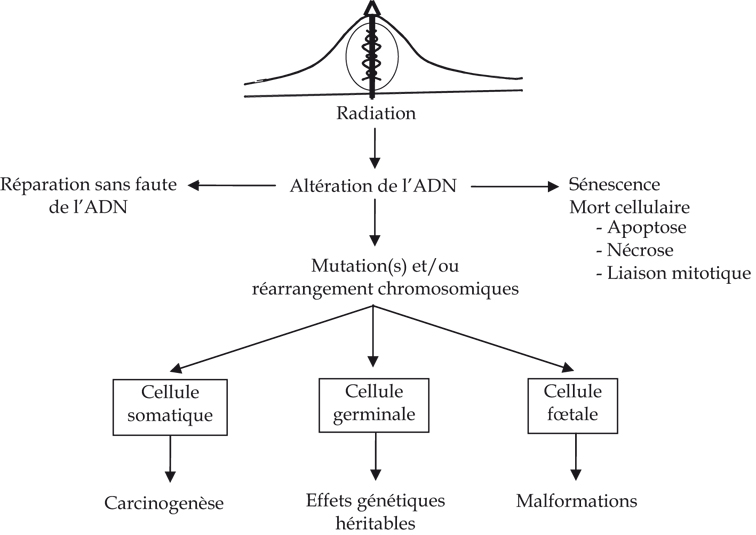
Cependant, durant les dernières décennies, de nombreux résultats indiquent que ce modèle n’est pas suffisant, et que d’autres effets « non ciblés » existent, y compris aux faibles doses et débits de dose (Little, 2006). Ces effets ont été mis en évidence par des études in vitro et in vivo. Ils mettent en œuvre des interactions cellulaires ou tissulaires. Plusieurs rapports récents ont fait la revue des connaissances sur ces différents effets (Gourmelon et coll., 2005; Tubiana et coll., 2005; National Research Council, 2005; Nénot et Sugier, 2006). Les paragraphes suivants en résument les principaux points.
). Ces effets ont été mis en évidence par des études in vitro et in vivo. Ils mettent en œuvre des interactions cellulaires ou tissulaires. Plusieurs rapports récents ont fait la revue des connaissances sur ces différents effets (Gourmelon et coll., 2005; Tubiana et coll., 2005; National Research Council, 2005; Nénot et Sugier, 2006). Les paragraphes suivants en résument les principaux points.Médiation intra-cytoplasmique
Des expériences in vitro reposent sur une irradiation très localisée d’une partie spécifique de la cellule à l’aide de micro-faisceaux. Ces expériences montrent qu’une irradiation cytoplasmique (ne touchant pas le noyau) peut induire des mutations de l’ADN, sans effet notable sur la survie des cellules. Ces résultats sous-tendent l’existence de médiateurs intra-cytoplasmiques impliqués dans l’induction de lésions ou dans la réparation de l’ADN.
Effet de proximité aux faibles doses (bystander effect)
Cet effet est défini comme l’induction d’effets biologiques dans des cellules qui n’ont pas été directement traversées par une particule chargée, mais qui se trouvaient à proximité des cellules touchées. Les effets induits peuvent être une augmentation de la fréquence de mutations ou une modification de l’expression de certains gènes. Plusieurs expérimentations ont permis de mettre en évidence ce phénomène :
• transfert de milieu de culture irradié : augmentation de la fréquence de mutations chez des cellules non irradiées placées dans un milieu de culture qui avait auparavant accueilli des cellules ayant reçu une irradiation alpha ;
• co-cultures : cultures de cellules sur les deux faces internes opposées d’une bouteille contenant un milieu de culture, la distance entre les deux faces étant supérieure au trajet d’une particule alpha. L’irradiation des cellules d’une face entraîne également une augmentation de la fréquence des mutations parmi les cellules de l’autre face (non irradiées) ;
• irradiation ciblée de cellules : irradiateur capable de bombarder des cellules avec des particules alpha avec une précision telle qu’il est possible de savoir exactement dans une culture cellulaire combien de cellules et lesquelles ont reçu une particule. Avec cet outil, les chercheurs ont pu montrer que les cellules voisines de cellules irradiées présentaient également une augmentation de la fréquence des mutations.
Ces résultats sous-tendent l’existence de médiateurs intra-cytoplasmiques, trans-membranaires et mêmes extra-cellulaires, capables d’induire une augmentation de la fréquence de mutations. Les effets de proximité n’ont été observés jusqu’à présent que pour les particules alpha et des micro-faisceaux de particules chargées, et à des doses faibles (lorsque peu de cellules sont traversées par une particule).
Réponse adaptative
Dès la fin des années 1990, certaines expérimentations ont montré que les effets d’une irradiation à forte dose d’une cellule (fréquence des mutations, remaniements chromosomiques…) étaient moins importants si cette cellule avait au préalable été soumise à une irradiation à faible dose. Ce phénomène de réponse adaptative semble être lié à une réduction du phénomène d’apoptose (mort cellulaire programmée). D’autres mécanismes pourraient agir via une stimulation du système de réparation de l’ADN. Aujourd’hui, il existe de nombreux résultats d’expérimentations animales montrant que des expositions prolongées à faibles débits peuvent induire une adaptation du système hématopoïétique. Il se produit alors une résistance relative. Ces expositions peuvent induire l’activation des fonctions immunitaires ; dans les mêmes conditions d’exposition, des effets inhibiteurs de la croissance des tumeurs malignes, du développement des métastases et de la carcinogenèse en général, peuvent être constatés.
Instabilité génomique
Dans des modèles expérimentaux de cellules soumises à des rayonnements ionisants, les chromosomes deviennent parfois instables plusieurs générations cellulaires après l’irradiation. Une telle instabilité est constatée également dans le vieillissement ou à la suite d’un stress. Ce phénomène engendre des remaniements qui peuvent conduire à la perte ou au gain de segments de chromosomes et entraîner des variations du nombre de certains gènes. Il contribue ainsi à démasquer certaines mutations récessives, auparavant silencieuses et participant à la cancérogenèse. Une instabilité génomique radio-induite a été mise en évidence in vivo sur des lymphocytes d’individus irradiés. Des résultats in vitro semblent indiquer que des signaux en provenance de cellules irradiées peuvent stimuler des réarrangements chromosomiques dans des cellules qui n’étaient pas présentes lors de l’irradiation.
Modulation du système immunitaire
À fortes doses, l’exposition aux rayonnements ionisants conduit souvent à une immuno-suppression. Néanmoins, en plus de cet effet cytotoxique, les rayonnements peuvent déclencher un « signal de danger » qui peut influencer la réponse cellulaire d’ordre immunitaire. Les rayonnements ionisants se comporteraient donc plutôt comme un modulateur de l’immunité. Ces mécanismes pourraient être impliqués dans le phénomène de réponse adaptative.
En résumé, de nombreux résultats ont été obtenus au cours des dernières décennies, tant sur les mécanismes de réparation des lésions et de la réponse cellulaire que sur ceux de l’instabilité génétique et de la transformation. Ces résultats permettent de comprendre certains paramètres qui régissent certaines étapes des processus de mutagenèse et de carcinogenèse. Toutefois, ces mécanismes sont encore insuffisamment connus pour autoriser une description générale en fonction de la nature des rayonnements, des doses et des débits de dose. De plus, les implications éventuelles de ces mécanismes au niveau d’un tissu ou d’un organisme sont aujourd’hui inconnues. L’impact de ces mécanismes sur la relation entre le risque de cancer et l’exposition aux rayonnements ionisants aux faibles doses et débits de dose reste un sujet de controverse (Brenner et Sachs, 2006; Tubiana et coll., 2006).
; Tubiana et coll., 2006).Bibliographie
[1] achanzar we, brambila em, diwan ba, webber mm, waalkes mp. Inorganic arsenite-induced malignant transformation of human prostate epithelial cells.
J Natl Cancer Inst. 2002;
94:1888- 1891
[2] adami ho, lipworth l, titus-ernstoff l, hsieh cc, hanberg a, et coll.. Organochlorine compounds and estrogen-related cancers in women.
Cancer Causes Control. 1995;
6:551- 566
[3] adamson iyr, bakowska j, bowden dh. Mesothelial cell proliferation after instillation of long or short asbestos fibers into mouse lung.
Amer J Pathol. 1993;
142:1209- 1216
[4] afonja o, raaka bm, huang a, das s, zhao x, et coll.. RAR agonists stimulate SOX9 gene expression in breast cancer cell lines: evidence for a role in retinoid-mediated growth inhibition.
Oncogene. 2002;
21:7850- 7860
[5] antila e, mussalo-rauhamaa h, kantola m, atroshi f, westermarck t. Association of cadmium with human breast cancer.
Sci Total Environ. 1996;
186:251- 256
[6] ayotte p, dewailly e, brisson j. L’exposition aux composés organochlorés estrogéniques et le cancer du sein.
Bull Info Sante Envir (Quebec). 1994;
5:1- 3
[7] barouki r, morel y. Repression of cytochrome P450 1A1 gene expression by oxidative stress: mechanisms and biological implications.
Biochem Pharmacol. 2001;
61:511- 516
[8] benassayag c, perrot-applanat m, ferre f. Phytoestrogens as modulators of steroid action in target cells.
J Chromatogr B Analyt Technol Biomed Life Sci. 2002;
777:233- 248
[9] bertazzi pa, zocchetti c, pesatori ac, guercilena s, sanarico m, et coll.. Ten-year mortality study of the population involved in the Seveso incident in 1976.
American Journal of Epidemiology. 1989;
129:1187- 1200
[10] bird mg, greim h, snyder r, rice jm. International symposium: Recent advances in benzene toxicity.
Chem Biol Interact. 2005;
153-154:1- 5
[11] blankenship a, matsumura f. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced activation of a protein tyrosine kinase, pp60src, in murine hepatic cytosol using a cell-free system.
Mol Pharmacol. 1997;
52:667- 675
[12] both k, henderson dw, turner dr. Asbestos and erionite fibres can induce mutations in human lymphocytes that result in loss of heterozygosity.
Int J Cancer. 1994;
59:538- 542
[13] both k, turner dr, henderson dw. Loss of heterozygosity in asbestos-induced mutations in a human mesothelioma cell line.
Environ Molecular Mutagenesis. 1995;
26:67- 71
[14] brama m, gnessi l, basciani s, cerulli n, politi l, et coll.. Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism.
Molecular and Cellular Endocrinology. 2007;
264:102- 108
[15] brenner dj, sachs rk. Estimating radiation-induced risks at very low doses: rationale for using a linear no-threshold approach.
Rad Environ Biophys. 2006;
44:253- 256
[16] bretveld rw, thomas cm, scheepers pt, zielhuis ga, roeleveld n. Pesticide exposure: the hormonal function of the female reproductive system disrupted?.
Reprod Biol Endocrinol. 2006;
4: 30p.
[17] calle ee, frumkin h, henley sj, savitz da, thun mj. Organochlorines and breast cancer risk.
CA: A Cancer Journal for Clinicians. 2002;
52:301- 309
[18] carlson db, perdew gh. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins.
J Biochem Mol Toxicol. 2002;
16:317- 325
[19] casper rf, quesne m, jolivet a, milgrom e, savouret jf. Resveratrol, a component of red wine, has antagonistic activity on the arylhydrocarbon receptor.
Mol Pharmacol. 1999;
56:784- 790
[20] chen cj, wang cj. Ecological correlation between arsenic level in well water and age-adjusted mortality from malignant neoplasms.
Cancer Res. 1990;
50:5470- 5474
[21] collins s, marletta ma. Carcinogen-binding proteins. High-affinity binding sites for benzo[a]pyrene in mouse liver distinct from the Ah receptor.
Mol Pharmacol. 1984;
26:353- 359
[22] coumoul x, barouki r. Génotoxicité des métabolites des oestrogènes et cancers.
Medecine/Sciences. 2002;
22:13- 17
[23] darro f, cahen p, vianna a, decaestecker c, nogaret jm, et coll.. Growth inhibition of human in vitro and mouse in vitro and in vivo mammary tumor models by retinoids in comparison with tamoxifen and the RU-486 anti-progestagen.
Breast cancer research and treatment. 1998;
51:39- 55
[24] davidson ne. Environmental estrogens and breast cancer risk.
Current Opinion in Oncology. 1998;
10:475- 478
[25] denison ms, nagy sr. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals.
Annual Review of Pharmacology and Toxicology. 2003;
43:309- 334
[26] diry m, tomkiewicz c, koehle c, coumoul x, bock kw, et coll.. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism.
Oncogene. 2006;
25:5570- 5574
[27] dixon d, bowser ad, badgett a, haseman jk, brody ar. Incorporation of bromodeoxyuridine (BrdU) in the bronchiolar-alveolar regions of the lungs following two inhalation exposures to chrysotile asbestos in strain A/J mice.
J Environ Pathol Toxicol Oncol. 1995;
14:205- 213
[28] dopp e, schiffmann d. Analysis of chromosomal alterations induced by asbestos and ceramic fibers.
Toxicol Lett. 1998;
96-97:155- 162
[29] dopp e, saedler j, stopper h, weiss dg, schiffmann d. Mitotic disturbances and micronucleus induction in syrian hamster embryo fibroblast cells caused by asbestos fibers.
Environ Health Perspect. 1995;
103:268- 271
[30] dopp e, schuler m, schiffmann d, eastmond da. Induction of micronuclei, hyperdiploidy and chromosomal breakage affecting the centric/pericentric regions of chromosomes 1 and 9 in human amniotic fluid cells after treatment with asbestos and ceramic fibers.
Mutat Res. 1997;
377:77- 87
[31] driscoll ke. Effects of fibres on cell proliferation, cell activation and gene expression.
In : Mechanisms of fibre carcinogenesis. In: kane ab, boffetta p, saracci r, wilbourn jd (eds), editors.
IARC Scientific Publications. 1999;
140:7398
[32] eastmond da, mondrala st, hasegawa l. Topoisomerase II inhibition by myeloperoxidase-activated hydroquinone: a potential mechanism underlying the genotoxic and carcinogenic effects of benzene.
Chem Biol Interact. 2005;
153-154:207- 216
[33] eisinger f, alby n, bremond a, dauplat j, espie m, et coll.. Recommendations for medical management of women with genetic risk of developing breast and/or ovarian cancer: the INSERM-FNCLCC ad hoc committee.
Annales de génétique. 1999;
42:51- 64
[34] eskenazi b, mocarelli p, warner m, chee wy, gerthoux pm, et coll.. Maternal serum dioxin levels and birth outcomes in women of Seveso, Italy.
Environmental Health Perspectives. 2003;
111:947- 953
[35] feige jn, gelman l, rossi d, zoete v, métivier r, et coll.. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis.
J Biol Chem. 2007;
282:19152- 19166
[36] frueh fw, hayashibara kc, brown po, whitlock jp jr. Use of cDNA microarrays to analyze dioxin-induced changes in human liver gene expression.
Toxicol Lett. 2001;
122:189- 203
[37] gouédard c, barouki r, morel y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism.
Mol Cell Biol. 2004;
24:5209- 5222
[38] gourmelon p, barbey p, barescut jc, bouville a, cancio d, et coll.. Les conséquences sanitaires des contaminations internes chroniques par des radionucléides. Avis sur le rapport CERI « Etudes des effets sanitaires de l’exposition aux faibles doses de radiations ionisantes à des fins de radioprotection » et recommandations de l’IRSN.
Institut de radioprotection et de sûreté nucléaire, Rapport DRPH n° 2005-20. IRSN. Fontenay aux Roses:2005;
http://www.irsn.org.
[39] hei tk, piao cq, he zy, vannais d, waldren ca. Chrysotile fiber is a strong mutagen in mammalian cells.
Cancer Res. 1992;
52:6305- 6309
[40] hirabayashi y. p53-dependent gene profiling for reactive oxygen species after benzene inhalation: special reference to genes associated with cell cycle regulation.
Chem Biol Interact. 2005;
153-154:165- 170
[41] huang c, ke q, costa m, shi x. Molecular mechanisms of arsenic carcinogenesis.
Mol Cell Biochem. 2004;
255:57- 66
[42]iarc. Benzene. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, vol. 7.
International agency for research on cancer. Lyon: France.
1974;
203221
[43] iskander k, jaiswal ak. Quinone oxidoreductases in protection against myelogenous hyperplasia and benzene toxicity.
Chem Biol Interact. 2005;
153-154:147- 157
[44] jaga k. What are the implications of the interaction between DDT and estrogen receptors in the body?.
Med Hypotheses. 2000;
54:18- 25
[45] jaurand mc, levy f. Effets cellulaires et moléculaires de l’amiante.
Med Sci. 1999;
15:1370- 1378
[46] jaurand mc. Mechanisms of fiber-induced genotoxicity.
Environ Health Perspect. 1997;
105:1073- 1084
[47] jaurand mc. Use of in-vitro genotoxicity and cell transformation assays to evaluate the potential carcinogenicity of fibres.
In : Mechanisms of fibre carcinogenesis. In: kane ab, boffetta p, saracci r, wilbourn jd (eds), editors.
IARC Scientific Publications. 1999;
140:5572
[48] jensen cg, jensen lcw, rieder cl, cole rw, ault jg. Long crocidolite asbestos fibers cause polyploidy by sterically blocking cytokinesis.
Carcinogenesis. 1996;
17:2013- 2021
[49] jensen cg, watson m. Inhibition of cytokinesis by asbestos and synthetic fibres.
Cell Biol Intl. 1999;
23:829- 840
[50] johnson md, kenney n, stoica a, hilakivi-clarke l, singh b, et coll.. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland.
Nat Med. 2003;
9:1081- 1084
[51] jordan vc, mittal s, gosden b, koch r, lieberman me. Structure-activity relationships of estrogens.
Environmental Health Perspectives. 1985;
61:97- 110
[52] kane ab. Mechanisms of mineral fibre carcinogenesis.
In : Mechanisms of fibre carcinogenesis. In: kane ab, boffetta p, saracci r, wilbourn jd (eds), editors.
IARC Scientific Publications. 1999;
140:1134
[53] key t, reeves g. Organochlorines in the environment and breast cancer.
BMJ (Clinical Research ed). 1994;
308:1520- 1521
[54] kogevinas m. Human health effects of dioxins: cancer, reproductive and endocrine system effects.
Hum Reprod Update. 2001;
7:331- 339
[55] kortenkamp a. Breast cancer, oestrogens and environmental pollutants: a re-evaluation from a mixture perspective.
International Journal of Andrology. 2006;
29:193- 198
[56] lacassagne a. Apparition de cancers de la mamelle chez, la souris mâle soumise à des injections de folliculine.
CR Acad Sci. 1932;
195:630- 632
[57] laden f, hunter dj. Environmental risk factors and female breast cancer.
Annual Review of Public Health. 1998;
19:101- 123
[58] lan q, zhang l, li g, vermeulen r, weinberg rs, et coll.. Hematotoxicity in workers exposed to low levels of benzene.
Science. 2004;
306:1774- 1776
[59] lechner jf, tokiwa t, laveck m, benedict wf, banks-schlegel s, et coll.. Asbestos-associated chromosomal changes in human mesothelial cells.
Proc Natl Acad Sci. 1985;
82:3884- 3888
[60] lecureur v, ferrec el, n’diaye m, vee ml, gardyn c, gilot d, fardel o. ERK-dependent induction of TNFalpha expression by the environmental contaminant benzo(a)pyrene in primary human macrophages.
FEBS Lett. 2005;
579:1904- 1910
[61] lewis dr, southwick jw, ouellet-hellstrom r, rench j, calderon rl. Drinking water arsenic in Utah: A cohort mortality study.
Environ Health Perspect. 1999;
107:359- 365
[62] liehr jg. Hormone-associated cancer: mechanistic similarities between human breast cancer and estrogen-induced kidney carcinogenesis in hamsters.
Environmental Health Perspectives. 1997;
105 (suppl 3):565- 569
[63] liehr jg. Genotoxicity of the steroidal oestrogens oestrone and oestradiol: possible mechanism of uterine and mammary cancer development.
Hum Reprod Update. 2001;
7:273- 281
[64] liehr jg, avitts ta, randerath e, randerath k. Estrogen-induced endogenous DNA adduction: possible mechanism of hormonal cancer.
Proc Natl Acad Sci USA. 1986;
83:5301- 5305
[65] little jb. Lauriston S Taylor lecture: non targeted effects of radiation: implications for low-dose exposures.
Health Phys. 2006;
91:416- 426
[66] marchand a, tomkiewicz c, marchandeau jp, boitier e, barouki r, garlatti m. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces insulin-like growth factor binding protein-1 gene expression and counteracts the negative effect of insulin.
Mol Pharmacol. 2005;
67:444- 452
[67] massaad c, barouki r. An assay for the detection of xenoestrogens based on a promoter containing overlapping EREs.
Environ Health Perspect. 1999;
107:563- 566
[68] matthews j, zacharewski t. Differential binding affinities of PCBs, HO-PCBs, and aroclors with recombinant human, rainbow trout (Onchorhynkiss mykiss), and green anole (Anolis carolinensis) estrogen receptors, using a semi-high throughput competitive binding assay.
Toxicol Sci. 2000;
53:326- 339
[69] mcelroy ja, shafer mm, trentham-dietz a, hampton jm, newcomb pa. Cadmium exposure and breast cancer risk.
J Natl Cancer Inst. 2006;
98:869- 873
[70] mcgavran pd, brody ar. Chrysotile inhalation induces tritiated thymisine incorporation by epithelial cells of distal bronchioles.
Am J Respir Cell Biol. 1989;
1:231- 235
[71] meplan c, mann k, hainaut p. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells.
J Biol Chem. 1999;
274:31663- 31670
[72] mitra ak, faruque fs, avis al. Breast cancer and environmental risks: where is the link?.
Journal of Environmental Health. 2004;
66:24- 32
[73] monchaux g, bignon j, jaurand mc, lafuma j, sebastien p, et coll.. Mesotheliomas in rats following inoculation with acid-leached chrysotile asbestos and other mineral fibres.
Carcinogenesis. 1981;
2:229- 236
[74] n’diaye m, le ferrec e, lagadic-gossmann d, corre s, gilot d, et coll.. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene.
J Biol Chem. 2006;
281:19906- 19915
[75]national research council (nrc). Board on Radiation Effects Research. Health risks from exposure to low levels of ionizing radiation.
BEIR, VII Report, phase II. Washington, DC: National Academy of Science. 2005;
[76]national toxicology program. Report on Carcinogens.
Ninth Edition; U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program. 2000;
[77] nénot jc, sugier a. Synthèse par l’IRSN des rapports de l’UNSCEAR - période 2003-2007.
Institut de Radioprotection et de Sûreté Nucléaire. Fontenay Aux Roses:Documents de Doctrine et Synthèse, IRSN 2006-74. Septembre 2006;
http://www.irsn.org.
[78] nesatyy vj, ammann aa, rutishauser bv, suter mj. Effect of cadmium on the interaction of 17beta-estradiol with the rainbow trout estrogen receptor.
Environmental Science & Technology. 2006;
40:1358- 1363
[79] ohtake f, baba a, takada i, okada m, iwasaki k, et coll.. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase.
Nature. 2007;
446:562- 566
[80] ohtake f, takeyama k, matsumoto t, kitagawa h, yamamoto y, et coll.. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor.
Nature. 2003;
423:545- 550
[81] park jy, shigenaga mk, ames bn. Induction of cytochrome P4501A1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin or indolo(3,2-b)carbazole is associated with oxidative DNA damage.
Proceedings of the National Academy of Sciences of the United States of America. 1996;
93:23222327
[82] perdew gh, hollenback ce. Analysis of photoaffinity-labeled aryl hydrocarbon receptor heterogeneity by two-dimensional gel electrophoresis.
Biochemistry. 1990;
29:6210- 6214
[83] pesatori ac, consonni d, bachetti s, zocchetti c, bonzini m, et coll.. Short- and long-term morbidity and mortality in the population exposed to dioxin after the “Seveso accident”.
Ind Health. 2003;
41:127- 138
[84] philipp r, hughes ao. Morbidity and soil levels of cadmium. .
International Journal of Epidemiology. 1982;
11:257- 260
[85] polkowski k, mazurek ap. Biological properties of genistein. A review of in vitro and in vivo data.
Acta Pol Pharm. 2000;
57:135- 155
[86] poser i, rahman q, lohani m, yadav s, becker hh, et coll.. Modulation of genotoxic effects in asbestos-exposed primary human mesothelial cells by radical scavengers, metal chelators and a glutathione precursor.
Mutat Res. 2004;
559:19- 27
[87] rivedal e, witz g. Benzene metabolites block gap junction intercellular communication. Role in hematotoxicity and leukemia?.
Chem Biol Interact. 2005;
153-154:257- 260
[88] rowlands jc, gustafsson ja. Aryl hydrocarbon receptor-mediated signal transduction.
Crit Rev Toxicol. 1997;
27:109- 134
[89] rusyn i, peters jm, cunningham ml. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver.
Crit Rev Toxicol. 2006;
36:459- 479
[90] safe s, mcdougal a. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for hormone-dependent cancers.
Int J Oncol. 2002;
20:1123- 1128
[91] safe sh, zacharewski t. Organochlorine exposure and risk for breast cancer.
Progress in Clinical and Biological Research. 1997;
396:133- 145
[92] savouret jf, antenos m, quesne m, xu j, milgrom e, et coll.. 7-ketocholesterol is an endogenous modulator for the Aryl hydrocarbon Receptor.
J Biol Chem. 2001;
276:3054- 3059
[93] setchell kd, zimmer-nechemias l, cai j, heubi je. Exposure of infants to phyto-oestrogens from soy-based infant formula.
Lancet. 1997;
350:23- 27
[94] setchell kd, zimmer-nechemias l, cai j, heubi je. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life.
The American Journal of Clinical Nutrition. 1998;
68:1453S- 1461S
[95] shukla a, gulumian m, hei tk, kamp d, rahman q, et coll.. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases.
Free Radic Biol Med. 2003;
34:1117- 1129
[96] smith mt, vermeulen r, li g, zhang l, lan q, et coll.. Use of ‘Omic’ technologies to study humans exposed to benzene.
Chem Biol Interact. 2005;
153-154:123- 127
[97] smith mt. The mechanism of benzene-induced leukemia: a hypothesis and speculations on the causes of leukemia.
Environ Health Perspect. 1996;
104:1219- 1225
[98] snedeker sm. Pesticides and breast cancer risk: a review of DDT, DDE, and dieldrin.
Environmental Health Perspectives. 2001;
109 (suppl 1):35- 47
[99] starek a. Estrogens and organochlorine xenoestrogens and breast cancer risk.
International Journal of Occupational Medicine and Environmental Health. 2003;
16:113- 124
[100] stoica a, katzenellenbogen bs, martin mb. Activation of estrogen receptor-alpha by the heavy metal cadmium.
Mol Endocrinol. 2000;
14:545- 553
[101] swaneck ge, fishman j. Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: characterization and intranuclear localization.
Proc Natl Acad Sci USA. 1988;
85:7831- 7835
[102] tapiero h, ba gn, tew kd. Estrogens and environmental estrogens.
Biomed Pharmacother. 2002;
56:36- 44
[103] tijet n, boutros pc, moffat id, okey ab, tuomisto j, pohjanvirta r. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries.
Mol Pharmacol. 2006;
69:140- 153
[104] tubiana m, aurengo a, averbeck d, bonnin d, leguen b, et coll.. Dose effect relationship and the estimation of the carcinogenic effects of low doses of ionising radiation.
Académie Nationale de Médecine, Institut de France-Académie des Sciences. Joint Report n° 2. March 30;
2005;
Edition Nucléon;
Paris:2005;
[105] tubiana m, aurengo a, averbeck d, masse r. Recent reports on the effects of low doses of ionising radiation and its dose-effect relationship.
Rad Environ Biophys. 2006;
44:245- 251
[106] upadhyay d, panduri v, ghio a, kamp dw. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria.
Am J Respir Cell Mol Biol. 2003;
29:180- 187
[107] van der meeren a, fleury j, nebut m, monchaux g, janson x, et coll.. Mesothelioma in rats following intrapleural injection of chrysotile and phosphorylated chrysotile (chrysophosphate).
Int J Cancer. 1992;
50:937- 942
[108] vaughan at, betti cj, villalobos mj, premkumar k, cline e, jiang q, diaz mo. Surviving apoptosis: a possible mechanism of benzene-induced leukemia.
Chem Biol Interact. 2005;
153-154:179- 185
[109] vermeulen r, lan q, zhang l, gunn l, mccarthy d, et coll.. Decreased levels of CXC-chemokines in serum of benzene-exposed workers identified by array-based proteomics.
Proc Natl Acad Sci USA. 2005;
102:17041- 17046
[110] wang ns, jaurand mc, magne l, kheuang l, pinchon mc, et coll.. The interactions between asbestos fibers and metaphase chromosomes of rat pleural mesothelial cells in culture. A scanning and transmission electron microscopic study.
Am J Pathol. 1987;
126:343- 349
[111] weiss c, faust d, durk h, kolluri sk, pelzer a, et coll.. TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway.
Oncogene. 2005;
24:4975- 4983
[112] widerak m, ghoneim c, dumontier mf, quesne m, corvol mt, et coll.. The aryl hydrocarbon receptor activates the retinoic acid receptoralpha through SMRT antagonism.
Biochimie. 2005;
88:387- 397
[113] wolff ms, weston a. Breast cancer risk and environmental exposures.
Environmental health perspectives. 1997;
105 (suppl 4):891- 896
[114] wormke m, stoner m, saville b, walker k, abdelrahim m, et coll.. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes.
Mol Cell Biol. 2003;
23:1843- 1855
[115] wu j, liu w, koenig k, idell s, broaddus vc. Vitronectin adsorption to chrysotile asbestos increases fiber phagocytosis and toxicity for mesothelial cells.
Am J Physiol Lung Cell Mol Physiol. 2000;
279:L916- L923
[116] yegles m, saint-etienne l, renier a, janson x, jaurand mc. Induction of metaphase and anaphase/telophase abnormalities by asbestos fibers in rat pleural mesothelial cells in vitro.
Amer J Respir Cell Mol Biol. 1993;
9:186- 191
[117] yegles m, janson x, dong hy, renier a, jaurand mc. Role of fibre characteristics on cytotoxicity and induction of anaphase/telophase aberrations in rat pleural mesothelial cells in vitro. Correlations with in vivo animal findings.
Carcinogenesis. 1995;
16:2751- 2758
[118] yoon bi, hirabayashi y, kawasaki y, kodama y, kaneko t, et coll.. Aryl hydrocarbon receptor mediates benzene-induced hematotoxicity.
Toxicol Sci. 2002;
70:150156
→ Aller vers le SOMMAIRE de l'ouvrage