| |
| Med Sci (Paris). 2008 November; 24(11): 901–903. Published online 2008 November 15. doi: 10.1051/medsci/20082411901.Comment les granzymes tuent leurs cibles Denis Martinvalet* and Jérôme Thiery Immune Disease Institute, Harvard Medical School, 200 Longwood Avenue, Boston MA 02115, États-Unis MeSH keywords: Apoptose, Caspases, Mort cellulaire, Cytoplasme, Granzymes, Mitochondries, Modèles biologiques |
Perforine, granulysine et granzymes, effecteurs de la cytotoxicité Les lymphocytes T cytotoxiques (CTL) et les cellules natural killer (NK) sont essentiels à la réponse immunitaire anti-tumorale et à la défense contre les pathogènes intracellulaires. Des études chez la souris et chez l’homme ont montré que l’élimination des cellules cancéreuses et des cellules infectées par des pathogènes intracellulaires tels que les mycobactéries et les virus implique l’exocytose des granules cytotoxiques de ces lymphocytes [
12]. En revanche, l’extinction de la réponse immunitaire nécessite l’activation de la voie Fas. Les granules cytotoxiques, des lysosomes de sécrétion spécialisés, contiennent la perforine (PFN) et la granulysine - deux protéines capables de former des pores dans la membrane plasmique et d’altérer la paroi des bactéries - ainsi qu’une famille de protéases à sérine appelées granzymes. Parmi les cinq granzymes décrites chez l’homme et les dix identifiées chez la souris, les granzymes A (GzmA) et B (GzmB) sont les plus abondantes. Une fois les cellules cibles reconnues, les CTL ou les cellules NK sécrètent le contenu de leurs granules dans la synapse immunologique. PFN permet l’entrée des Gzm dans la cellule cible, déclenchant ainsi des cascades biochimiques multiples qui, bien que distinctes, conduisent toutes à la mort cellulaire [
1,
2]. |
Comment les Granzymes tuent leurs cibles : une nouvelle voie apoptotique pour GzmA Au cours des dernières décennies, d’intenses recherches ont permis de mieux comprendre les mécanismes apoptotiques initiés par GzmB. Comme les caspases, GzmB clive ses substrats au niveau de résidus acide aspartiques, et initie ainsi la perforation de la membrane externe de la mitochondrie, libérant des protéines pro-apoptotiques (cytochrome c, HtrA2/Omi, endo G, Smac/Diablo et AIF, apoptosis-inducing factor) depuis l’espace inter-membranaire de la mitochondrie vers le cytoplasme. Suivant les cas, cette voie est dépendante ou indépendante des caspases [1, 2]. L’essentiel des travaux portant sur la voie de la mort cellulaire induite par une autre granzyme, GzmA, a été effectué par l’équipe du Pr. Judy Lieberman. Au sein de cette équipe, nous avons montré que la mort cellulaire induite par GzmA présente toutes les caractéristiques morphologiques de l’apoptose, c’est-à-dire la condensation de la chromatine, la fragmentation de l’ADN, l’externalisation des phosphatidyl-sérines à la surface cellulaire, la perte du potentiel de membrane (ΔΨm) mitochondriale et la production de ROS (reactive oxygen species) [
13]. Cependant, cette voie est complètement indépendante des caspases et des protéines anti-apoptotiques de la famille Bcl2. Dans cette nouvelle voie apoptotique, la fragmentation de l’ADN se fait par cassures simple brin. Par ailleurs, nous avons établi que le complexe SET est une cible-clé de GzmA [
3]. Ce complexe, qui est associé au réticulum endoplasmique, comprend les nucléases activées par GzmA, NM23-H1 et TREX 1, la protéine phosphatase PP2A et son inhibiteur pp23, et trois substrats directs de GzmA, les protéines Set, Ape1 et HMGB2 impliquées respectivement dans l’assemblage des nucléosomes, la réparation des altérations de l’ADN induites par le stress oxydatif et le remodelage de la chromatine. Lors de l’apoptose induite par GzmA, la translocation rapide du complexe SET vers le noyau conduit à la dégradation de la protéine Set par GzmA, libérant ainsi l’endonucléase NM23-H1 qui, de concert avec l’exonucléase TREX 1, induit la fragmentation de l’ADN [
4,
5]. En même temps, GzmA clive et inactive Ku70, HMGB2, Ape 1, empêchant ainsi la réparation de l’ADN [
6–
8]. |
Tous les chemins pro-apoptotiques de GzmA et GzmB convergent vers la mitochondrie La mitochondrie tient une place centrale dans l’apoptose induite par GzmA. Dans les minutes suivant le traitement de cellules avec GzmA et PFN, nous avons observé une forte production de ROS accompagnée d’une perte du ΔΨm mitochondrial. GzmA agit directement sur la mitochondrie puisque la production de ROS et la perte du ΔΨm sont obtenues par simple addition de GzmA à des mitochondries purifiées, en l’absence de facteurs cytosoliques. Nous avons aussi démontré que les ROS produites par la mitochondrie en réponse à GzmA, et en particulier l’anion superoxyde (
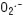 ), sont non seulement indispensables pour la translocation nucléaire du complexe SET mais aussi pour la mort cellulaire induite par GzmA et par les CTL. De plus, la production de ROS et la perte du ΔΨm ne sont pas accompagnées de la libération de protéines pro-apoptotiques de l’espace inter-membranaire de la mitochondrie vers le cytoplasme. Contrairement à ce qui se passe lors de l’apoptose induite par GzmB et Fas-L, la membrane mitochondriale externe demeure intacte lors de l’apoptose induite par GzmA. Nos travaux démontrent aussi pour la première fois que la production des ROS est indépendante de l’altération de la membrane mitochondriale externe et que ces ROS sont nécessaires à la mort cellulaire induite par GzmA [
9] (Figure 1). Afin de comprendre les mécanismes moléculaires d’induction de ces ROS par GzmA, nous avons utilisé une approche protéomique à partir de mitochondries purifiées. Nous avons ainsi identifié NDUFS3 (la sous unité 30 kDa du complexe I NADH : Ubiquinone oxydoréductase de la chaîne respiratoire) comme étant une cible de GzmA. GzmA hydrolyse NDUFS3 et inhibe l’activité enzymatique du complexe I, conduisant ainsi à la production de ROS et à la mort cellulaire. En effet, des cellules surexprimant une forme non-hydrolysable de NDUFS3 ne produisent plus de ROS et deviennent résistantes à l’apoptose induite soit par GzmA soit par l’attaque de CTL n’exprimant que GzmA. Par ailleurs, l’équipe du Dr Douglas Green a démontré que la caspase 3 clive NDUFS1, la sous unité de 75 kDa de ce même complexe I de la chaîne respiratoire [
10]. Ces résultats montrent donc que des voies apoptotiques différentes convergent vers le complexe I, soulignant ainsi l’importance de son inactivation et de la production de ROS au cours de la mort cellulaire. ), sont non seulement indispensables pour la translocation nucléaire du complexe SET mais aussi pour la mort cellulaire induite par GzmA et par les CTL. De plus, la production de ROS et la perte du ΔΨm ne sont pas accompagnées de la libération de protéines pro-apoptotiques de l’espace inter-membranaire de la mitochondrie vers le cytoplasme. Contrairement à ce qui se passe lors de l’apoptose induite par GzmB et Fas-L, la membrane mitochondriale externe demeure intacte lors de l’apoptose induite par GzmA. Nos travaux démontrent aussi pour la première fois que la production des ROS est indépendante de l’altération de la membrane mitochondriale externe et que ces ROS sont nécessaires à la mort cellulaire induite par GzmA [
9] (Figure 1). Afin de comprendre les mécanismes moléculaires d’induction de ces ROS par GzmA, nous avons utilisé une approche protéomique à partir de mitochondries purifiées. Nous avons ainsi identifié NDUFS3 (la sous unité 30 kDa du complexe I NADH : Ubiquinone oxydoréductase de la chaîne respiratoire) comme étant une cible de GzmA. GzmA hydrolyse NDUFS3 et inhibe l’activité enzymatique du complexe I, conduisant ainsi à la production de ROS et à la mort cellulaire. En effet, des cellules surexprimant une forme non-hydrolysable de NDUFS3 ne produisent plus de ROS et deviennent résistantes à l’apoptose induite soit par GzmA soit par l’attaque de CTL n’exprimant que GzmA. Par ailleurs, l’équipe du Dr Douglas Green a démontré que la caspase 3 clive NDUFS1, la sous unité de 75 kDa de ce même complexe I de la chaîne respiratoire [
10]. Ces résultats montrent donc que des voies apoptotiques différentes convergent vers le complexe I, soulignant ainsi l’importance de son inactivation et de la production de ROS au cours de la mort cellulaire.  | Figure 1.
Mécanismes d’induction de la mort cellulaire par Granzyme A. Dans le cytosol de la cellule cible GzmA est transporté dans la matrice mitochondriale probablement par le complexe TIM/TOM. GzmA clive NDUFS3 au sein du complexe I afin de produire des ROS. Ces ROS permettent la translocation nucléaire du complexe SET où GzmA hydrolyse Set permettant aux nucléases NM23-H1 et TREX de fragmenter l’ADN afin d’induire la mort cellulaire. |
|
Comment GzmA pénètre t-elle dans la mitochondrie ? Tout n’est pas résolu, en particulier, comment GzmA peut-elle atteindre son substrat NDUFS3 de localisation mitochondriale ? En effet, GzmA ne possède pas de séquence d’adressage mitochondrial et n’altère pas la membrane mitochondriale externe. Un début de réponse nous vient de l’observation que tous les effets de GzmA sur la mitochondrie sont dépendants du ΔΨm mitochondrial. En effet, la valinomycine, un protonophore qui annule le ΔΨm mitochondrial, inhibe l’hydrolyse de NDUFS3 et la production des ROS induite par GzmA. Nous avons aussi démontré que GzmA interagit de façon spécifique avec les protéines Hsp70 et Hsp90. En plus de leur fonction de chaperon, ces protéines sont des cofacteurs de la machinerie d’import protéique de la mitochondrie. Cette machinerie utilise le ΔΨm comme force motrice afin de faire entrer les pré-protéines dans l’organite. Il est donc probable que GzmA utilise cette voie d’import protéique afin de pénétrer dans la matrice mitochondriale. |
Parce que la mort cellulaire activée par GzmA est indépendante des caspases et de Bcl2, elle semble être un atout du système immunitaire pour lutter contre les cellules cancéreuses surexprimant Bcl2 et contre des cellules infectées par des virus exprimant des inhibiteurs de caspases. Par ailleurs, une étude récente a démontré que le complexe SET est impliqué dans l’altération de l’ADN et l’apoptose des neurones lors d’accidents vasculaires cérébraux. Dans cette étude, les auteurs ont démontré qu’en réponse à une chute du pH intracellulaire, l’endopeptidase lysosomale à arginine (AEP) clive Set et libère NM23-H1 afin d’induire la fragmentation de l’ADN [
11]. Les souris déficientes en AEP sont résistantes à ce type de mort neuronale. Il en découle que l’étude de la mort cellulaire induite par GzmA aidera non seulement à une meilleure compréhension de l’apoptose induite par les cellules immunitaires mais aussi celle induite par le stress des neurones. De façon plus générale, ces travaux devraient faciliter l’élaboration de nouvelles approches pour le traitement de tumeurs, de maladies auto-immunes ou neuro-dégénératives. |
Nous remercions le Dr Edouard Vannier et le Dr Emmanuel Derudder pour la relecture de ce texte.
|
1. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 2008; 26 : 389–420. 2. Lieberman J, Fan Z. Nuclear war: the granzyme A-bomb. Curr Opin Immunol 2003; 15 : 553–9. 3. Beresford PJ, Zhang D, Oh DY, et al. Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem 2001; 276 : 43285–93. 4. Fan Z, Beresford PJ, Oh DY, et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003; 112 : 659–72. 5. Chowdhury D, Beresford PJ, Zhu P, et al. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 2006; 23 : 133–42. 6. Fan Z, Beresford PJ, Zhang D, et al. HMG2 interacts with the nucleosome assembly protein SET and is a target of the cytotoxic T-lymphocyte protease granzyme A. Mol Cell Biol 2002; 22 : 2810–20. 7. Fan Z, Beresford PJ, Zhang D, et al. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol 2003; 4 : 145–53. 8. Zhu P, Zhang D, Chowdhury D, et al. Granzyme A, which causes single-stranded DNA damage, targets the double-strand break repair protein Ku70. EMBO Rep 2006; 7 : 431–7. 9. Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 2005; 22 : 355–70. 10. Martinvalet D, Dykxhoorn DM, Ferrini R, et al. Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 2008; 133 : 681–92. Liu Z, Jang SW, Liu X, et al. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell 2008; 29 : 665–78. 12. Ménasché G, Ménager M, Le Deist F, Fischer A, de Saint Basile G. Défaut d’exocytose des granules lytiques : plusieurs causes, un même effet. Med Sci (Paris) 2006; 22 : 733–8. 13. Barouki R. Stress oxydant et vieillissement. Med Sci (Paris) 2006; 22 : 266–72. |


