| |
| Med Sci (Paris). 2007 May; 23(5): 526–532. Published online 2007 May 15. doi: 10.1051/medsci/2007235526.La granulomatose septique chronique X+
Un fabuleux modèle d’étude de l’activation du complexe NADPH oxydase Marie-José Stasia* Centre de diagnostic et de recherche sur la granulomatose septique chronique, GREPI, TIMC/Imag UMR CNRS 5525, laboratoire d’Enzymologie, BP 217, 38043 Grenoble Cedex 09, France |
La granulomatose septique chronique (CGD) est une maladie héréditaire orpheline, survenant avec une fréquence de 1/250 000 individus, répertoriée comme un syndrome d’immunodéficience héréditaire. Elle se caractérise cliniquement par des infections bactériennes ou fongiques graves et récidivantes, survenant dès le plus jeune âge. Cette maladie peut être diagnostiquée à l’âge adulte, bien que rarement. La CGD est la conséquence du dysfonctionnement du complexe NADPH (nicotinamide adenine dinucleotide phosphate) oxydase des cellules phagocytaires (neutrophiles, éosinophiles, monocytes, macrophages), normalement responsable d’une consommation intense d’oxygène (O2) à l’origine de la production de dérivés toxiques encore appelés formes réactives de l’oxygène (FRO, ou ROS, reactive oxygen species anglophones). Ces dérivés issus de l’activation des phagocytes sont pourvus de pouvoirs bactéricide et fongicide. La CGD doit son nom à la présence de foyers inflammatoires de type granulomateux dans différents parenchymes, constitués de cellules géantes, multinucléées, résultant de la fusion de macrophages ayant séquestré des bactéries non détruites [
1]. |
La granulomatose septique chronique, aspects moléculaires Le complexe NADPH oxydase Le complexe NADPH oxydase se compose d’un élément protéique membranaire, le cytochrome b
558 formé de deux sous-unités p22 phox et gp91 phox ( phox vient de phagocyte oxidase, la gp91 phox est encore appelée Nox2), et de facteurs cytosoliques p47 phox, p67 phox et p40 phox. Récemment, six homologues de gp91 phox appartenant à la famille Nox-Duox ont été identifiés. Leur distribution tissulaire et leur régulation sont très différentes suggérant une grande variété de fonctions physiologiques. La modulation de leur activité peut entraîner des évènements physiopathologiques très divers [
2]. Lorsque le neutrophile est au repos, le complexe NADPH oxydase est dissocié et inactif. Lors de la reconnaissance spécifique d’un élément pathogène par la cellule phagocytaire, une cascade de signalisation intracellulaire est déclenchée, aboutissant à la phosphorylation des facteurs cytosoliques. Ces éléments cytosoliques p47phox, p67phox et p40phox migrent ensuite vers la membrane plasmique où ils s’associent au cytochrome b
558, formant un complexe NADPH oxydase actif. À partir de l’oxygène moléculaire (O2), ce complexe est alors capable de produire des ions superoxyde
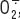 , point de départ de la synthèse de FRO bactéricides (Figure 1). Des petites protéines G, Rac1/2 et Rap1 ainsi que des protéines liant le calcium (MRP8 et MRP14) sont également impliquées dans la régulation de l’activité du complexe oxydase [2–
4]. , point de départ de la synthèse de FRO bactéricides (Figure 1). Des petites protéines G, Rac1/2 et Rap1 ainsi que des protéines liant le calcium (MRP8 et MRP14) sont également impliquées dans la régulation de l’activité du complexe oxydase [2–
4].  | Figure 1.
Mécanismes bactéricides du neutrophile. L’enzyme responsable de la production d’anions superoxyde
à partir de la réduction de l’oxygène moléculaire, est la NADPH oxydase. Il s’agit d’un système de transfert d’électrons depuis le NADPH (issu du shunt des pentoses) vers le FAD, puis jusqu’à l’oxygène moléculaire qui est réduit en . Les ions superoxyde produits sont instables et aboutissent à différents dérivés bactéricides comme le peroxyde d’hydrogène H 2O 2 produit par dismutation spontanée ou en présence de superoxyde dismutase leucocytaire, ou les ions hypochlorites ClO − , puissants oxydants. Lors de l’explosion respiratoire, d’importantes quantités de formes réactives de l’O 2 vont être produites dans la vacuole de phagocytose. Le passage d’électrons à travers la membrane vacuolaire dû à l’activation du complexe NADPH oxydase, est électrogénique et s’accompagne d’un processus de compensation de charges (H + et/ou K +). |
Gp91phox, élément redox principal du complexe oxydase La grosse sous unité du cytochrome b
558 (gp91 phox ou Nox2) est une glycoprotéine de 570 acides aminés et de masse moléculaire 91 kDa. C’est l’élément redox de l’oxydase. Le profil d’hydrophobicité de la protéine indique la présence de 6 hélices transmembranaires situées du côté amino-terminal comportant 3 sites extracellulaires de glycosylation [
5]. Le modèle généralement admis comporte une séquence amino-terminale cytosolique, deux boucles B et D intracytoplasmiques et une queue cytosolique carboxy-terminale contenant les sites potentiels de fixation du FAD (flavine adénine dinucléotide) et du NADPH (Figure 2). Leur localisation a été déterminée par comparaison avec des séquences de flavoenzymes de la famille des ferrédoxines réductases (FNR) [
6]. Le cytochrome b
558 est porteur de deux hèmes liés à quatre histidines situées dans le troisième et le cinquième passage transmembranaire de gp91 phox. L’existence d’un canal à protons, situé au niveau du troisième passage transmembranaire de gp91 phox, permettrait la compensation de charge engendrée par le passage des électrons au travers de la membrane plasmique lors de l’activation du complexe oxydase. Cette théorie a été controversée ; gp91 phox ne serait pas un canal à proton mais servirait à moduler d’autres canaux préexistants [
7,
8].  | Figure 2.
Localisation des mutations du gène CYBB à l’origine des cas de CGD de type X+. La protéine gp91phox se compose d’une séquence amino-terminale comprenant six passages transmembranaires, trois boucles extracellulaires (A, C et E) sur lesquelles sont situés trois sites de glycosylation, deux boucles intra-cytoplasmiques (B et D) et une séquence carboxy-terminale cytosolique comportant les sites potentiels de fixation du NADPH et du FAD. Gp91phox comporte deux hèmes liés aux histidines figurées en noir sur le schéma et localisées au niveau du troisième et du cinquième passage transmembranaire. Les mutations humaines à l’origine des CGD X+ sont principalement ponctuelles et faux-sens, localisées préférentiellement dans la partie cytosolique carboxy-terminale de la protéine gp91phox. Les mutations figurées en rouge sont celles qui ont fait l’objet d’une modélisation dans la lignée cellulaire PLB-985 dont le gène CYBB a été préalablement invalidé. |
Les différentes formes génétiques de la CGD Il existe deux formes de transmission de la CGD : l’une est liée à l’X (CGDX), c’est la forme la plus fréquente, l’autre est de type autosomique récessif (CGDAR) (Figure 3). Des mutations dans le gène CYBB codant gp91 phox et localisé sur le bras court du chromosome X (Xp21.1), sont à l’origine de la CGDX. Il existe trois formes de CGDAR selon le gène affecté, NCF1, NCF2 ou CYBA codant respectivement les protéines p47 phox, p67 phox et p22 phox. Hormis celle qui implique un défaut génique quasiment constant du gène NCF1 (une délétion GT au début de l’exon 2), ces formes sont très rares. Cela est probablement dû à des recombinaisons chromosomiques avec un pseudogène (Ψ NCF1) contenant cette mutation, extrêmement homologue au gène NCF1 [
9]. Généralement les mutations géniques n’ont pas de localisation spécifique (région promotrice des gènes, jonctions intron/exon, exons), et tous les types de mutations (délétions et/ou insertions de taille variable avec décalage du cadre de lecture, mutations ponctuelles non-sens, faux sens) peuvent être rencontrées [ 9–
11].  | Figure 3.
Les différentes formes génétiques de la granulomatose septique chronique CGD. La CGD résulte de mutations dans quatre gènes distincts : CYBB, CYBA, NCF1 et NCF2 codant respectivement les protéines gp91 phox, p22 phox, p47 phox et p67phox du complexe NADPH oxydase des neutrophiles [ 9]. Selon le taux d’expression de la protéine gp91 phox, la forme CGD liée à l’X peut être classifiée en trois variants différents X 0, X − et X +. L’encart correspond à l’immunodétection de gp91 phox dans un extrait détergent de neutrophiles de patients atteints de ces différents variants de CGD [ 11]. Les pourcentages indiqués entre parenthèses représentent la fréquence des types de CGD retrouvés, par exemple la CGD lié à l’X de type X 0 représente 55 % de tous les cas de CGD décrits. |
La plupart des mutations géniques conduisent à l’absence de la protéine correspondante, soit par un défaut de synthèse de l’ARN messager, soit par une instabilité de la protéine mutée (tronquée ou mal conformée) rapidement éliminée (CGDX0 ou CGDAR0). La forme dite CGDX0, la plus classique, se traduit par une absence des deux sous-unités du cytochrome b
558, bien que seul le gène CYBB soit atteint. Il semblerait qu’il existe une stabilisation mutuelle de ces deux sous-unités, car dans les CGDAR avec une déficience en p22phox, gp91phox est également absente (Figure 3). L’expression clinique des CGD liées à l’X est généralement plus sévère que celle des formes de transmission autosomique récessive [1, 9]. Il existe des variants rares caractérisés soit par une sous expression (CGDX−), soit par une expression normale de gp91phox mutée (CGDX+). Les variants CGDX− résultent de mutations faux-sens à l’origine d’une perte de la conformation stable de gp91phox ou plus rarement, de mutations dans le promoteur du gène CYBB [
12]. Les mutations faux-sens dans la partie codante du gène sont principalement la cause des CGDX+. La rareté de ces formes variantes est illustrée par les résultats d’une étude récente répertoriant les différentes mutations à l’origine de CGDX publiées dans la littérature internationale. Sur 191 mutations, on dénombre 150 cas de CGDX0, 26 cas de CGDX− et seulement 17 cas de CGDX+ [
13]. |
Modélisation cellulaire des variants CGD de type X + pour l’étude des processus d’activation du complexe oxydase Conséquences fonctionnelles des mutations de gp91phox à l’origine des CGD X+
La CGD, et plus particulièrement les formes X +, constitue un formidable modèle d’étude du complexe NADPH oxydase. L’étude de ces variants permet d’établir un lien direct entre une région et une fonction spécifique de la protéine gp91 phox. Cela constitue une ouverture fascinante pour la compréhension des mécanismes d’activation et d’assemblage du complexe oxydase. Les mutations à l’origine des CGDX+ sont principalement localisées dans la partie carboxy-terminale cytosolique de gp91phox (Figure 2). Cela suggère que cette séquence est impliquée majoritairement dans le fonctionnement de l’oxydase mais intervient peu dans la stabilisation de la structure protéique [
14]. Sur les 18 mutations répertoriées dans la littérature, seules 8 ont fait l’objet d’une étude fonctionnelle. Des régions de gp91phox impliquées directement ou indirectement dans l’interaction des facteurs cytosoliques p47phox et/ou p67phox lors de l’assemblage du complexe oxydase, ont été découvertes grâce à l’étude des polynucléaires neutrophiles de patients atteints de CGDX+. La partie carboxy-terminale cytosolique de gp91phox semble particulièrement impliquée dans ce processus, puisque les mutations His303Asn/Pro304Arg, Cys369Arg, Gly408Glu, Asp500Gly et Glu568Lys inhibent la translocation des facteurs cytosoliques vers la membrane plasmique des neutrophiles des patients étudiés, après l’activation du complexe NADPH oxydase. De plus le transfert des électrons du NADPH vers le FAD est très souvent perturbé chez ces patients [
15–
19]. L’étude du variant CGDX+ dû à la mutation Pro415His a permis de confirmer la localisation du site potentiel de fixation du NADPH et notamment le motif G-X-G-X-X-P essentiel à la liaison de la partie pyrophosphate du NADPH [6, 19]. Il est à noter que la mutation Gly408Glu située dans ce site, entraîne également l’inhibition de l’assemblage du complexe oxydase. Cela indique que la fixation du NADPH sur son site pourrait faciliter une conformation de gp91phox favorable à une interaction avec les facteurs cytosoliques p47phox et/ou p67phox. L’importance du site de fixation du FAD (338HPFT341) dans le maintien d’une activité NADPH oxydase a également été soulignée par la découverte de la mutation Thr341Lys d’un mutant CGDX+ [
18]. La mutation Arg54Ser de gp91phox, une des rares mutations localisée dans la partie membranaire de gp91phox, près de l’ancrage du premier hème, affecte les propriétés spectrales du cytochrome b
558, ainsi que le passage des électrons du FAD vers les hèmes [15]. Cela confirme le modèle structural de gp91phox et conforte l’idée que les deux hèmes sont essentiels au passage électronique du FAD vers l’O2 extracellulaire. Étude de l’activation du complexe NADPH oxydase par modélisation des cas de CGDX+ dans la lignée promyélocytaire PLB-985 Bien que l’étude fonctionnelle des polynucléaires neutrophiles des patients CGDX + apporte certaines informations structurales et fonctionnelles, la modélisation cellulaire des mutations humaines est nécessaire pour l’obtention d’un matériel biologique suffisamment abondant afin d’aborder la compréhension des mécanismes d’activation du complexe oxydase au niveau moléculaire. Le modèle cellulaire le plus performant pour cette approche est sans doute la lignée promyélocytaire PLB-985 établie en 1987 par l’équipe du Pr Tom Rado à partir de cellules myéloïdes d’une patiente atteinte de leucémie aiguë [
20]. La différenciation des cellules de cette lignée en polynucléaires neutrophiles est facilement induite. En 1993, l’équipe du Pr Mary Dinauer a établi une lignée PLB-985 dans laquelle le gène CYBB codant gp91phox a été inactivé par recombinaison homologue (PLB-985 KO)[
21]. Il est donc possible, à partir de cette lignée, d’obtenir en culture des neutrophiles de type CGDX0. L’approche expérimentale consiste à reproduire les mutations humaines dans l’ADNc de gp91phox par mutagenèse dirigée. Après clonage de l’ADNc muté dans un vecteur d’expression eucaryote, celui-ci est transfecté de façon stable dans les cellules PLB-985 KO par électroporation. Après sélection des clones positifs, le suivi d’expression de la protéine gp91phox mutée se fait par immunodétection ou par cytométrie de flux à l’aide d’anticorps spécifiques. En 1999, l’équipe du Pr M. Dinauer a réalisé la première étude fonctionnelle, dans la lignée PLB-985, d’une mutation humaine (délétion 488-497) à l’origine d’une CGDX+ [
22,
23]. Cette mutation se situe dans une région particulièrement importante de la queue carboxy-terminale cytosolique de gp91phox (Thr483 à Gln507) (Figure 2), qui correspond à une boucle en hélice présente dans la séquence de tous les analogues Nox-Duox [2,
24]. Cette séquence pourrait intervenir dans le processus de régulation de l’assemblage du complexe oxydase tout en contrôlant l’accès du NADPH à son site de liaison (Figure 4). Au repos, l’hélice α recouvrirait le site de fixation du NADPH afin d’en interdire l’accès. Lors de l’activation de la NADPH oxydase, les facteurs cytosoliques s’associeraient avec gp91phox entraînant un changement conformationnel de cette protéine avec ouverture du site de fixation du NADPH par le glissement de l’hélice α à l’extérieur de ce site [
25]. La délétion 488-497, bien qu’amputant une partie de l’hélice α ne semble pas empêcher la translocation des facteurs cytosoliques à la membrane plasmique lors de l’activation du complexe oxydase, mais elle inhibe le transfert d’électrons du NADPH vers le FAD. Cependant, les auteurs n’ont pas déterminé si le défaut d’activité oxydase associé à cette mutation était dû à une mauvaise conformation du cytochrome b
558 et/ou à un défaut de fixation du NADPH à son site.  | Figure 4.
Représentation graphique tridimensionnelle de la queue cytosolique de gp91
phox. La séquence carboxy-terminale de gp91phox (bleu) vue à partir de l’intérieur de la membrane cellulaire, comporte les sites de fixation du FAD et du NADPH qui sont enchâssés dans une crevasse (A). Au repos, l’accès du site de fixation du NADPH semble obstrué par le recouvrement de l’hélice α (beige) composée des résidus Thr483-Gln507. La représentation (B) est obtenue après une rotation de 90° de la représentation en (A) autour de l’axe de la membrane. Agrandissement de l’hélice α à partir de la représentation en (B) et positionnement des acides aminés jouant un rôle stratégique dans l’activité du complexe NADPH oxydase (C).
|
La boucle en hélice α de la partie cytosolique carboxy-terminale de gp91phox comporte des acides aminés chargés qui semblent jouer un rôle décisif dans les processus d’assemblage et d’activation du complexe oxydase. La mutation Asp500Gly, dont l’impact fonctionnel a été préalablement étudié à partir des neutrophiles d’un patient atteint de CGDX+ [
17], a été reproduite dans le modèle PLB-985, permettant ainsi de démontrer l’importance de la charge négative de cet acide aminé. En effet, l’annulation ou la réversion de sa charge négative (Asp500Gly, Asp500Ala, Asp500Arg) a un effet drastique sur l’activité NADPH oxydase ainsi que sur la translocation des facteurs cytosoliques [
26]. Un effet identique est obtenu par l’annulation ou l’inversion de la charge d’autres acides aminés chargés localisés dans cette hélice α cytosolique (Asp484Thr His490Asp) (Figure 4). La mutation Leu505Arg de gp91phox, caractéristique d’un cas de CGDX+ récemment diagnostiqué au laboratoire (Figure s 2 et 4), a été également modélisée dans les cellules PLB-985 [11,
27]. Cette leucine est totalement conservée dans les protéines Nox-Duox, suggérant un rôle fonctionnel commun dans cette famille. Le cytochrome b
558 recombinant muté a été purifié afin d’étudier les mécanismes moléculaires d’assemblage du complexe oxydase in vitro, en présence des protéines p47phox, p67phox et Rac1 recombinantes. La mutation Leu505Arg influence l’interaction de p67phox avec le cytochrome b
558. Des concentrations trois fois plus importantes de p67phox sont nécessaires pour l’activation maximum du turn-over du cytochrome b
558 muté. De plus, l’affinité du cytochrome b
558 muté purifié pour le NADPH est trois fois plus importante que celle du cytochrome b
558 sauvage. Cela indique que la liaison du NADPH à son site est probablement étroitement liée à l’interaction du cytochrome b
558 avec p67phox. La mutation Leu505Arg pourrait perturber les mouvements de l’hélice α et sa liaison avec p67phox, empêchant une accessibilité optimale du NADPH au niveau de son site de fixation. Il faut noter que par comparaison avec les séquences des membres de la famille FNR, la Leu505 fait partie du site potentiel de fixation de l’adénine du NADPH [25]. Cependant la plupart des FNR (ferredoxine-NADP+ réductase ne possèdent pas l’hélice α cytosolique dont l’insertion semble détruire la structure de cette partie du site de liaison du NADPH [
28,
29]. Enfin le modèle cellulaire PLB-985 KO a également permis d’étudier l’impact d’une double mutation His303Asn/Pro304Arg sur l’activité NADPH oxydase des neutrophiles d’un patient atteint de CGDX+ [
16]. Chaque mutation, His303Asn et Pro304Arg, ainsi que la double mutation, ont été reproduites et étudiées dans le modèle PLB-985 KO. Ce travail a permis d’exclure la possibilité que l’une des mutations soit un polymorphisme du gène CYBB, car chacune séparément entraîne un défaut d’assemblage du complexe oxydase à l’origine de l’inhibition de son activité [
30]. Il semble donc que la modélisation cellulaire de mutations humaines à l’origine de CGD X+ permette d’aborder les mécanismes moléculaires d’activation du complexe oxydase des phagocytes. |
Conclusionset perspectives L’étude du dysfonctionnement du complexe oxydase dans la CGD est une approche prometteuse pour la compréhension du fonctionnement de cet enzyme. La modélisation cellulaire dans la lignée PLB-985 KO des cas de CGDX+ ainsi que la production de mutants dans des régions spécifiques de gp91phox, permettent l’obtention d’un matériel biologique suffisamment fiable et abondant pour aborder l’étude moléculaire de l’activation du complexe oxydase. De la même manière, la modélisation des variants CGDX−, caractérisés par une expression réduite de la protéine gp91phox mutée, pourra être envisagée afin d’appréhender les mécanismes de synthèse et de maturation de gp91phox. Le modèle PLB-985 KO permet aussi l’étude de nouvelles régions de gp91phox dont le rôle fonctionnel est à ce jour inconnu, en créant des mutations de novo par mutagenèse dirigée. Récemment, grâce à cette approche, des mutants de la boucle D de gp91phox
(Figure 2) possédant une activité 5 à 10 fois supérieure à l’activité détectée dans des cellules de type sauvage, ont pu être mis en évidence [26]. La détermination des mécanismes moléculaires à l’origine de ces super-activités NADPH oxydase, permettront une avancée certaine dans la compréhension des processus d’activation/désactivation du complexe oxydase. Les données fonctionnelles et structurales obtenues grâce à cette approche méthodologique, permettront une meilleure compréhension du mode d’activation du complexe NADPH oxydase ainsi que la maîtrise des processus de synthèse de gp91phox et du cytochrome b
558.

|
L’auteur tient à remercier très sincèrement le Pr F. Morel, directeur de l’équipe GREPI, du laboratoire TIMC-Imag, UMR CNRS 5525, pour son soutien constant dans ce projet, les doctorantes Xing Jun Li et Clara Bionda, les techniciennes Cécile Martel, Michelle Guillot, Elisabeth Maquet, ayant participé au travail, les chercheurs ayant contribué activement aux résultats présentés, Franck Fieschi du laboratoire des Protéines Membranaires, Institut de Biologie Structurale, UMR 5075 CEA/CNRS/Université Joseph Fourier, Didier Grunwald du Département Réponse et Dynamique Cellulaire/CEA et Jacques Mathieu du Département de Radiobiologie, CRSSA, Grenoble, France. Le GREPI bénéficie du support financier de l’Université Joseph Fourier, Faculté de Médecine; la Région Rhône-Alpes, programme Émergence; le Ministère de l’Éducation et de la Recherche, MENRT ; la Direction de la Recherche Régionale Clinique, DRRC ; le Laboratoire Merck-Sharp Dohme-Chibret, le programme conjoint de recherche Tempra/Mira 2001-Région Rhône-Alpes et l’US Immunodeficiency Network (USIDNET) consortium du NIH, USA.
|
1. Winkelsteinw JA, Marino MC, Johnston RB, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000; 79 : 155–69. 2. Guichard C, Pedruzzi E, Fay M, et al. The Nox/Duox family of ROS-generating NADPHoxidases. Med Sci (Paris) 2006; 22 : 953–9. 3. Berthier S, Paclet MH, Lerouge S, et al. Changing the conformation state of cytochrome b558 initiates NADPH oxidase activation: MRP8/MRP14 regulation. J Biol Chem 2003; 278 : 25499–508. 4. Reeves EP, Lortat Jacobs HL, Messina CGM, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002; 416 : 291–7. 5. Wallach TM, Segal AW. Analysis of glycosylation sites on gp91phox, the flavocytochrome of the NADPH oxidase, by site-directed mutagenesis and translation in vitro. Biochem J 1997; 321 : 583–5. 6. Segal AW, West I, Wientjes F, et al. Cytochrome b245 is a flavocytochrome containing FAD and NADPH-binding site of the microbicidal oxidase of phagocytes Biochem J 1992; 284 : 781–8. 7. Henderson LM, Thomas S, Banting G, Chappell JB. The arachidonate-activatable, NADPH oxidase-associated H+ channel is contained within the multi-membrane-spanning N-terminal region of gp91-phox. Biochem J 1997; 325 : 701–5. 8. DeCoursey TE, Cherny VV, Morgan D, Katz BZ, Dinauer MC. The gp91phox component of NADPH oxidase is not the voltage-gated proton channel in phagocytes, but it helps. J Biol Chem 2001; 276 : 36063–6. 9. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000; 79 : 170–200. 10. Roos D, De Boer M, Kuribayashi F, et al. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood 1996; 87 : 1663–81. 11. Stasia MJ, Bordigoni P, Floret D, et al. Characterization of six novel mutations in the CYBB gene leading to different sub-types of X-linked chronic granulomatous disease. Hum Genet 2005; 116 : 72–82. 12. Stasia MJ, Brion JP, Boutonnat J, Morel F. Severe clinical forms of cytochrome b-negative chronic granulomatous disease (X91-) in 3 brothers with a point mutation in the promoter region of CYBB. J Infect Dis 2003; 188 : 1593–604. 13. Heyworth PG, Curnutte JT, Rae J, et al. Hematologically important mutations: X-linked chronic granulomatous disease (second update). Blood Cells Mol Dis 2001; 27 : 16–26. 14. Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol 2003; 15 : 578–84. 15. Cross AR, Heyworth PG, Rae J, Curnutte JT. A variant X-linked chronic granulomatous disease patient (X91+) with partially functional cytochrome b. J Biol Chem 1995; 270 : 8194–8200. 16. Stasia MJ, Lardy B, Maturana A, et al. Molecular and functional characterization of a new X-linked chronic granulomatous disease variant (X91+) case with a double missense mutation in the gp91-phox-cytosolique C-terminal tail. Biochem Biophys Acta 2002; 1586 : 316–30. 17. Leusen JH, de Boer M, Bolscher BG, et al. A point mutation in gp91-phox of cytochrome b558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47-phox and p67-phox. J Clin Invest 1994; 93 : 2120–6. 18. Leusen JH, Meischl C, Eppink MH, et al. Four novel mutations in the gene encoding gp91-phox of human NADPH oxidase : consequences for oxidase assembly. Blood 2000; 95 : 666–73. 19. Dinauer MC, Curnutte JT, Rosen H, Orkin SH. A missense mutation in the neutrophil cytochrome b heavy chain in cytochrome-positive X-linked chronic granulomatous disease. J Clin Invest 1989; 84 : 2012–6. 20. Tucker KA, Lilly MB, Heck L, Rado TA. Characterization of a new human diploid myeloid leukaemia cell line (PLB-985). Blood 1987; 70 : 372–8. 21. Zhen L, King AA, Xiao Y, et al. Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc
Natl Acad Sci USA 1993; 90 : 9832–6. 22. Schapiro BL, Newburger PE, Klempner MS, Dinauer MC. Chronic granulomatous disease presenting in a 69-year-old man. N Engl J Med 1991; 325 : 1786–90. 23. Yu L, Cross AR, Zhen L, Dinauer MC. Functional analysis of NADPH oxidase in granulocytic cells expressing a delta488-497 gp91(phox) deletion mutant. Blood 1999; 94 : 2497–504. 24. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4 : 181–9. 25. Taylor WR, Jones DT, Segal AW. A structural model for the nucleotide binding domains of the flavocytochrome b-245 beta-chain. Protein Sci 1993; 2 : 1675–85. 26. Li, XJ, Grunwald D, Mathieu J, Morel F, Stasia MJ. Crucial role of two potential cytosolic regions of Nox2, 191TSSTKTIRRS200 and 484DESQANHFAVHHDEEKD500, on NADPH oxidase activation. J Biol Chem 2005; 280 : 14962–73. 27. Li XJ, Fieschi F, Paclet MH, et al. Leu505 of Nox2 is crucial for optimal p67phox-dependent activation of the flavocytochrome b558 during phagocytic NADPH oxidase assembly. J Leukoc Biol 2007; 81 : 238–49. 28. Shatwell KP, Dancis A, Cross AR, Klausner RD, Segal AW. The FRE1 ferric reductase of Saccharomyces cerevisiae is a cytochrome b similar to that of NADPH oxidase. J Biol Chem 1996; 271 : 14240–4. 29. Karplus PA, Daniels MJ, Herriott JR. Atomic structure of ferredoxin-NADP+ reductase : prototype for a structurally novel flavoenzyme family. Science 1991; 251 : 60–6. 30. Bionda C, Li XJ, van Bruggen R, et al. Functional analysis of a two-amino acid substitution in gp91phox in a patient with X-linked flavocytochrome-b558-positive chronic granulomatous disease by means of transgenic PLB-985 cells. Hum Genet 2004; 115 : 418–27. |





