| |
| Med Sci (Paris). 2005 June; 21(6-7): 601–607. Published online 2005 June 15. doi: 10.1051/medsci/2005216-7601.Repliement des protéines : Études in vitro
Jeannine Yon-Kahn* Institut de biochimie, biophysique moléculaire et cellulaire, UMR CNRS, Université de Paris- Sud, 91405 Orsay, France |
Le repliement des protéines est un processus fondamental au cours duquel l’information contenue dans l’ADN est traduite en information tridimensionnelle, condition nécessaire à l’émergence des propriétés fonctionnelles. Ce processus complexe, qui représente l’étape ultime de traduction du message génétique (→), peut être considéré comme le premier acte de la morphogenèse. Comme l’a écrit Jacques Monod [
1] : « Dans le processus de structuration d’une protéine globulaire, on peut voir à la fois l’image microscopique et la source du développement épigénétique de l’organisme lui-même ». (→) m/s 2005, n° 6-7, p.563)
Les mécanismes impliqués dans le repliement des protéines ont intrigué les scientifiques dès le début du XXe siècle. La progression des études fut marquée par le contexte scientifique du moment, par l’état des connaissances et des techniques. Les concepts ont évolué dans différentes directions pour aboutir aujourd’hui à une vision unifiée de ce processus complexe. Les principes fondamentaux du repliement des protéines ont des applications pratiques, dans la compréhension des diverses pathologies liées aux repliements incorrects, dans l’exploitation des avancées de la recherche génomique et dans la conception de nouvelles protéines. Ainsi, le décryptage des mécanismes de repliement des protéines représente aujourd’hui un des principaux défis en biologie. C’est un domaine très actif où la frontière entre physique et biologie s’est estompée. |
À partir de 1930 émergèrent les premières études démontrant la réversibilité du processus de dénaturation. La première analyse significative décrivant les relations entre l’état natif et l’état dénaturé fut publiée en 1931 par Hsien Wu [
2]. Cette époque fut marquée par le succès dans la renaturation de plusieurs protéines. De ces premières études émergea le concept de réversibilité de la dénaturation inférant que le repliement est un processus spontané. Il resta cependant admis, pendant plusieurs décennies, que les protéines possédant des ponts disulfure ne pouvaient se renaturer qu’à la condition que ceux-ci n’aient pas été rompus. En 1945, Anson [
3] souligna le caractère de « tout ou rien » du processus de dénaturation-renaturation, rationalisé plus tard par la théorie des deux états. Entre 1950 et 1960, de très nombreux travaux furent consacrés à la thermodynamique des interactions non covalentes impliquées dans la stabilité de la structure native des protéines. Une attention particulière fut portée au rôle de l’eau ; en 1959, Kauzmann [
4] montra que l’effet hydrophobe constituait le facteur principal dirigeant le repliement des protéines, les acides aminés non polaires ayant tendance à s’enfouir à l’intérieur de la protéine et les acides aminés polaires à se localiser à la surface en contact avec le solvant. Cette conception, établie à partir de données thermodynamiques, fut confirmée ultérieurement lorsque les premières structures tridimensionnelles de protéines apparurent. L’aboutissement des études structurales par diffraction des rayons X à partir de 1960, puis par RMN à partir de 1980, offrait une nouvelle base pour les études de repliement. Ces aspects historiques ont été détaillés par Ghélis et Yon [
5]. |
Postulat d’Anfinsen et paradoxe de Levinthal À partir des années 60 commença une nouvelle ère pour les études de repliement des protéines. Cette période fut marquée par un événement capital : en 1961, Anfinsen et ses collègues [
6] démontrèrent que la ribonucléase complètement dénaturée, et dont les quatre ponts disulfure avaient été rompus, était capable de se replier spontanément en une enzyme complètement active, alors que la probabilité de formation des quatre ponts disulfure corrects était quasiment nulle. De plus, lorsque quelques ponts disulfure incorrects étaient formés initialement, ils se réarrangeaient au cours du repliement en présence d’un agent réducteur adéquat ou d’une enzyme, la protéine disulfure isomérase. C’est donc le repliement de la protéine qui dirige l’appariement correct des ponts disulfures. Anfinsen déduisit de ses résultats que « All the information necessary to achieve the native conformation of a protein in a given environment is contained in its amino acid sequence » [
7]1. Avec cette assertion, connue sous le nom de postulat d’Anfinsen, une nouvelle étape était franchie dans l’évolution des idées sur le repliement des protéines. Ce succès fut suivi d’un renouveau d’intérêt pour ce domaine de recherche. Le repliement d’une chaîne polypeptidique étant un processus spontané sous le contrôle de la séquence, il était admis comme corollaire au postulat d’Anfinsen que la structure native d’une protéine est la structure la plus stable dans un environnement donné. Cette conception fut remise en question par Levinthal en 1968, à partir de considérations temporelles [
8]. Considérant une chaîne polypeptidique de 100 acides aminés, et admettant l’hypothèse minimaliste que chacun d’eux peut adopter seulement deux conformations, on dénombre 1030 conformations possibles pour la chaîne entière. Si la conversion d’une conformation dans l’autre requiert seulement 10−11 seconde, une recherche au hasard de la conformation native à travers toutes les conformations possibles nécessiterait 1011 années, plus que le temps de l’univers ! Or in vivo comme in vitro, le repliement s’effectue en quelques secondes, au plus en quelques minutes. Il devenait donc évident que l’évolution avait trouvé une solution efficace pour résoudre ce problème combinatoire. Connue sous le nom de paradoxe de Levinthal, cette question a peu attiré l’attention à l’époque, mais a par la suite dominé les discussions pendant près de trente ans. |
Cette période fut suivie d’un renouveau d’intérêt pour les études de repliement. Les méthodes expérimentales s’étaient perfectionnées et permettaient de pénétrer dans le détail de la structure. L’analyse nécessitait l’existence de critères incontestables de l’état initial et de l’état final : si l’état natif se définit aisément lorsque la protéine possède une activité biologique, l’état totalement dénaturé est quant à lui plus difficile à caractériser. Tanford [
9] apporta une contribution importante à la caractérisation de l’état dénaturé et montra que seul le chlorure de guanidinium à forte concentration était capable de dénaturer totalement les protéines. La plupart des études de repliement des protéines étaient effectuées dans des conditions d’équilibre thermodynamique. En raison de la réversibilité du processus et du caractère abrupt des courbes de transition ainsi obtenues, caractère qui n’était pas sans rappeler les changements de phase solide-liquide, la dénaturation-renaturation fut interprétée selon la théorie des deux états. Cette interprétation consistait à admettre l’existence de deux seuls états macroscopiques, l’état natif et l’état dénaturé. L’application de la théorie des deux états permettait de déterminer les paramètres thermodynamiques du processus de repliement et d’évaluer en termes d’énergie libre la stabilité de la protéine native : les valeurs obtenues variaient entre – 5 et – 15 kcal/mol pour l’ensemble des protéines. Analysant la thermodénaturation du chymotrypsinogène sur la base de la théorie des deux états, Brandts [
10] montra en 1964 que la stabilité de la protéine dominée par les interactions hydrophobes suivait une loi parabolique en fonction de la température et était optimale à 10 °C. De part et d’autre de cette température, la stabilité de la protéine décroissait rapidement. Ce résultat fut à l’origine des études de dénaturation par le froid, qui apparurent cependant beaucoup plus tard. En fait, la température de stabilité optimale diffère pour chaque protéine. La validité de l’application de la théorie des deux états au processus de dénaturation-repliement des protéines fut l’objet de nombreuses discussions et controverses. L’un des critères utilisés lors des études à l’équilibre fut la coïncidence des courbes de transition obtenues à partir de plusieurs observables indépendantes. Ce critère est encore retenu aujourd’hui pour vérifier la validité de l’approximation des deux états, signifiant que, si des états intermédiaires existent, il ne sont pas significativement peuplés dans les conditions d’équilibre. L’identité des paramètres énergétiques déterminés à partir des courbes de transition à l’équilibre avec ceux obtenus directement par des mesures calorimétriques fut également utilisée comme critère de validité de la théorie des deux états. Dans ce but, Privalov et ses collaborateurs, à l’Université de Pushino, près de Moscou, entreprirent en 1971 la construction d’un microcalorimètre différentiel à balayage d’une grande sensibilité. Analysant par cette méthode la thermodénaturation de cinq petites protéines globulaires, la ribonucléase, le lysozyme, la chymotrypsine, le cytochrome c et la myoglobine, Privalov et Kheschinashvili [
11] observèrent que la déviation par rapport à un processus à deux états ne dépassait pas 5 % ; cette déviation fut attribuée à l’existence d’espèces intermédiaires très instables. Les données obtenues dans les conditions d’équilibre montraient que le processus de dénaturation-renaturation pouvait être décrit par une transition réversible, symétrique et très coopérative et que, au moins pour les petites protéines, l’approximation des deux états se trouvait fréquemment validée. |
Différents modèles de repliement furent proposés soit à partir d’observations expérimentales, soit à partir de considérations théoriques ou de simulations. Le modèle classique de nucléation-propagation, qui s’applique aux transitions hélice → pelote statistique, implique une étape de nucléation suivie d’une propagation rapide, l’étape limitante étant le processus de nucléation. Proposé initialement par Baldwin pour expliquer les résultats concernant la cinétique de repliement de la ribonucléase A, ce modèle fut abandonné à la lumière de nouveaux résultats. Plus récemment, Fersht [
12] présenta un modèle de nucléation-condensation selon lequel se formeraient des noyaux de structures locales stabilisés par des interactions à longue distance. Un modèle séquentiel et hiérarchique fut soutenu par plusieurs auteurs [
13,
14] et généralement accepté pendant plusieurs années. Dans ce modèle, le premier événement, la nucléation, est suivi par la formation rapide de structures secondaires (Encadré) qui s’associent pour former les structures supersecondaires, puis les domaines et finalement la protéine active, suivant un chemin unique. Pour les protéines oligomériques, l’étape ultime concerne l’association des sous-unités. Un modèle très semblable, le modèle de la charpente (framework model) [
15], implique la formation de structures secondaires dans une étape précoce du repliement, avant la mise en place de la structure tertiaire. Un modèle modulaire fondé sur l’examen des structures tridimensionnelles de protéines considère les domaines, et même les sous-domaines, comme des unités autonomes de repliement. Ce modèle accepte pour chaque module un chemin de repliement unique et hiérarchique [
16]. Ces modèles privilégient le rôle des interactions à courte distance dans les premiers événements du repliement. Le modèle de diffusion-collision, énoncé par Karplus et Weaver, en 1976, sur des bases théoriques, fut réévalué en 1994 [
17] à la lumière des données expérimentales alors disponibles. Il admet que la nucléation se produit simultanément en différentes parties de la chaîne polypeptidique, engendrant des microstructures qui diffusent, s’associent et deviennent coalescentes pour former des sous-structures ayant une conformation native. Ces microstructures ont des temps de vie contrôlés par la diffusion des segments, de sorte que le repliement d’une chaîne de 100 à 200 acides aminés peut se produire en des temps très courts, inférieurs à la seconde. Selon ce modèle, le repliement se produit par une série d’étapes de diffusion-collision. Le modèle de l’effondrement hydrophobe (hydrophobic collapse) admet que le premier événement du repliement consiste en la formation d’une espèce compacte, via des interactions hydrophobes à longue distance, qui se produit avant ou en même temps que l’établissement des structures secondaires [
18]. Il est inspiré de l’effet hydrophobe proposé par Kauzmann. Le modèle du jigzaw puzzle fut introduit par Harrison et Durbin en 1985 [
19]. Il implique l’existence de multiples chemins de repliement, comme dans l’assemblage des pièces d’un puzzle où il existe plusieurs voies pour aboutir à une solution unique. Selon ce modèle, l’identification des espèces intermédiaires représente une description cinétique plutôt que structurale, chaque intermédiaire consistant en espèces hétérogènes en équilibre rapide. Ce modèle, qui présente quelques similitudes avec celui de Karplus et Weaver, a été sujet de controverses pendant une dizaine d’années. |
Détection des intermédiaires de repliement Les tentatives de résolution du paradoxe de Levinthal furent le point de départ d’une floraison d’études cinétiques à partir des années 70. Pour la plupart des protéines, les cinétiques de dénaturation étaient monophasiques, alors que les cinétiques de repliement étaient biphasiques, voire multiphasiques, avec une ou plusieurs phases rapides suivies d’une phase lente. Aux premières études cinétiques, il convient d’associer les noms de Baldwin, de Tanford et de leurs collaborateurs. Ces études démontraient l’existence d’intermédiaires de repliement même lorsque l’approximation des deux états était validée dans les études à l’équilibre. Afin de déterminer la position des espèces intermédiaires dans le chemin de repliement, un formalisme rigoureux des cinétiques à multiples étapes fut développé par Tanford et Ikai [
20] sur la base d’un seul état natif et d’un seul état dénaturé de la protéine. Un formalisme différent fut ultérieurement présenté par Hagermann [
21] pour rendre compte des résultats obtenus par Baldwin sur le repliement de la ribonucléase : celui-ci avait en effet observé l’existence de deux formes de protéine dénaturée se repliant avec des cinétiques différentes. La phase lente du processus fut attribuée à l’isomérisation cis-trans des prolines qui furent ultérieurement identifiées. Toutefois, pour beaucoup d’autres protéines, la phase lente ne pouvait s’expliquer par l’isomérisation des prolines.
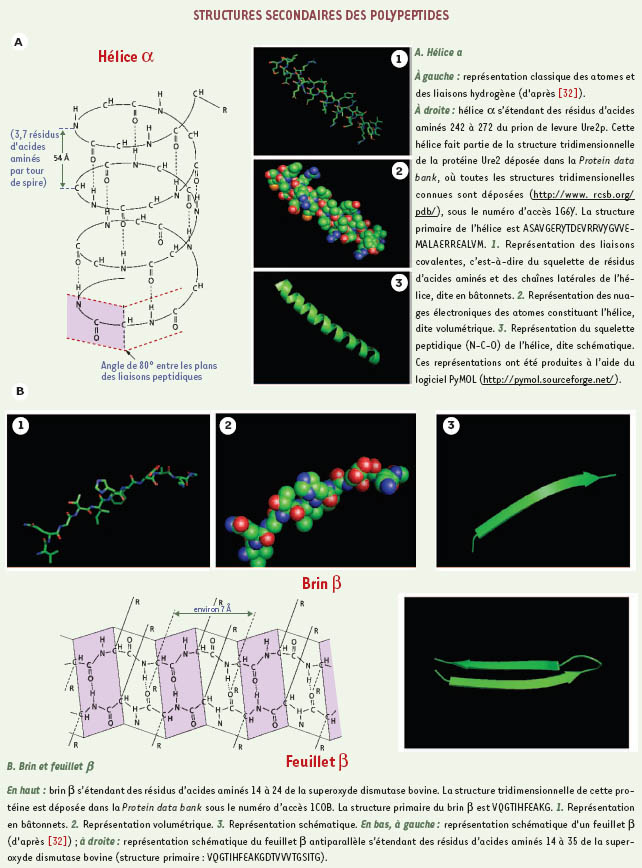
Caractérisation des intermédiaires de repliement La caractérisation structurale des espèces transitoires est essentielle à la compréhension des mécanismes de repliement. Elle se heurte à deux difficultés majeures, qui sont la coopérativité et la rapidité du processus, certains événements se produisant dans des temps inférieurs à la milliseconde ; les espèces transitoires ont des durées de vie très courtes. Malgré ces difficultés, de nombreux efforts ont été déployés pour caractériser les intermédiaires de repliement. Diverses stratégies ont été utilisées, telles que le blocage ou l’accumulation des espèces transitoires à bas pH, ou par l’ingénierie génétique, le marquage chimique rapide ou encore l’étude de fragments. Ces études ont bénéficié des avancées technologiques : les techniques de cinétique rapide, telles que les méthodes de flux ( stopped-flow) avec des constantes de temps de l’ordre de la milliseconde, couplées à des méthodes physiques (fluorescence, dichroïsme circulaire, puis RMN) ont été largement utilisées. Plus récemment, sont apparues des méthodes ultrarapides avec des constantes de temps de l’ordre de la microseconde et même de la nanoseconde. Ainsi, l’intervention de structures secondaires dans les étapes précoces du repliement a été observée pour la plupart des protéines [
22]. Ces espèces repliées et flexibles, dont l’existence avait été proposée par Ptitsyn et Rashin [15] à partir de considérations théoriques, ont été dénommées globule fondu (molten globule) par Ogushi et Wada [
23]. Ptitsyn et ses collègues [
24] en ont généralisé la notion, suggérant que le globule fondu est un intermédiaire obligatoire dans le repliement de toutes les protéines. Afin de surmonter une certaine confusion sémantique présente dans la littérature, Goldberg et al. [
25] ont introduit la notion de globule fondu spécifique : cette espèce représente un intermédiaire relativement compact ayant un contenu important de structures secondaires natives, mais une structure tertiaire fluctuante. Elle possède des surfaces hydrophobes accessibles à un colorant hydrophobe, l’anilinonaphtalène sulfonate (ANS). Dans cet intermédiaire, les résidus aromatiques conservent leur liberté de rotation, comme l’indique le spectre de dichroïsme circulaire dans le proche UV. Toutefois, pour certaines protéines, l’apparition de structures secondaires non natives a été détectée dans les étapes précoces du repliement ; une réorganisation des structures secondaires se produit alors dans des étapes ultérieures. Une abondante littérature concernant le globule fondu est apparue dans les années 90 (pour revue, voir [
26]). Selon une autre hypothèse, le tout premier événement du repliement d’une chaîne polypeptidique serait un effondrement hydrophobe, précédant la formation de structures secondaires ou se produisant simultanément. Plusieurs études expérimentales sont en accord avec ce mécanisme. L’existence de microstructures résiduelles constituées d’amas hydrophobes a été observée lors de la dénaturation de plusieurs protéines qui représenteraient des centres de nucléation pour le repliement. Enfin, des espèces oligomériques transitoires ont également été observées au cours du repliement de certaines protéines [
27]. Cinétiques du repliement Les espèces transitoires ainsi détectées apparaissant dans le temps mort d’un appareil de stopped-flow, qui est de l’ordre de la milliseconde, il n’est pas exclu que leur formation soit précédée d’événements encore plus précoces. Des avancées technologiques récemment développées dans des laboratoires spécialisés ont permis de meilleures résolutions temporelles, de l’ordre de la microseconde et même de la nanoseconde. Elles reposent principalement sur des méthodes de relaxation thermique utilisant des lasers [
28]. L’application de ces techniques a montré que la limite supérieure pour la vitesse de repliement d’une protéine est de l’ordre de la microseconde, en accord avec les études théoriques. La formation d’une hélice s’effectue en 10– 7 s, celle d’une boucle de dix résidus en 10– 6 s. L’effondrement hydrophobe initial de l’apomyoglobine en un état compact est terminé en moins de 20 µs : une première phase se produisant en 240 ns consiste en un effondrement local qui implique des contacts non natifs et la formation d’hélices ; dans une seconde phase durant 5 µs, l’une des hélices forme un contact avec deux autres hélices ; enfin, la protéine native apparaît dans une phase finale de 0,9 s. À la lumière de différentes études, il semble que les événements initiaux très rapides du repliement consistent en un effondrement hydrophobe accompagné ou non de la formation de structures secondaires natives ou non natives ; le globule fondu se forme après ces événements précoces. |
Une nouvelle vision du repliement des protéines : le paysage énergétique et l’entonnoir de repliement Pendant longtemps, la plupart des expérimentateurs ont accepté le modèle séquentiel du repliement avec une compétition cinétique entre le repliement correct et un repliement engendrant la formation d’agrégats (Figure 1).  | Figure 1.
Modèle séquentiel de repliement. Ce modèle envisage une compétition cinétique entre le repliement correct et un repliement engendrant la formation d’agrégats. |
Les progrès les plus décisifs ont résulté de la convergence des études théoriques, amorcées vers 1980, et des approches expérimentales. En 1995, un modèle unifié de repliement fondé sur la surface d’énergie effective de la protéine a émergé des études théoriques. Introduit par Wolynes et al. [
29], il représente le processus de repliement en termes de paysage énergétique et d’entonnoir de repliement, et décrit le comportement thermodynamique d’un ensemble de molécules dépliées jusqu’à l’état natif (Figure 2). Dans cette nouvelle vision, le repliement des protéines procède par l’organisation progressive d’ensembles de structures partiellement repliées, suivant de multiples chemins dépendant des paramètres énergétiques et des conditions. Le nombre de conformations diminue depuis le sommet jusqu’au bas de l’entonnoir, en même temps que décroît l’entropie de la chaîne polypeptidique ; le repliement est d’autant plus rapide que la pente est forte. Le paysage énergétique présente l’allure d’un entonnoir dont les aspérités des parois font apparaître des minima énergétiques locaux capables de « piéger » des espèces transitoires. Selon la hauteur des barrières d’énergie locales, les populations de molécules reprendront avec retard un chemin de repliement ou s’agrégeront. Les représentations tridimensionnelles de la Figure 3 montrent que, lorsque les parois de l’entonnoir sont lisses, le repliement s’effectue selon l’approximation des deux états ; dans l’autre cas, en revanche, des intermédiaires sont peuplés sur les chemins de repliement.  | Figure 2.
L’entonnoir de repliement. La largeur de l’entonnoir représente l’entropie de la chaîne polypeptidique, la hauteur l’énergie. Q est la fraction de contacts natifs pour chaque collection d’états. |
 | Figure 3.
Représentation tridimensionnelle d’entonnoirs de repliement. À gauche : les parois sont lisses et la pente est forte, le repliement se comporte selon l’approximation des deux états ; il n’y a pas de minima locaux. À droite : l’entonnoir fait apparaître de nombreux minima locaux ; les états intermédiaires sont peuplés et peuvent s’accumuler (d’après [
31]). |
La nouvelle vision se présente donc comme une rationalisation énergétique du modèle de jigsaw puzzle. Elle est validée actuellement par de nombreux résultats expérimentaux qui démontrent l’existence de populations hétérogènes sur le chemin de repliement. |
La compréhension des mécanismes de repliement des protéines représente l’un des principaux enjeux de la biologie. Il s’agit en effet d’un problème essentiel pour l’analyse des divers événements impliqués dans la régulation cellulaire, ainsi que pour l’utilisation de l’information contenue dans le nombre croissant de gènes séquencés (→) (→) m/s 2005, n° 6-7, p.609). L’élucidation de ces mécanismes devrait permettre le développement de nouvelles stratégies thérapeutiques pour la prévention et le traitement des maladies associées à des repliement incorrects. |
Footnotes |
1. Monod J. Le hasard et la nécessité. Paris : Seuil, 1976. 2. Wu H. Studies on denaturation of proteins: a theory of denaturation. Chin J Physiol 1931; 3 : 321–44. 3. Anson ML. Denaturation of proteins and properties of protein groups.
Adv Protein Chem 1945; 2 : 121–282. 4. Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem 1959; 14 : 1–67. 5. Ghélis C, Yon JM. Protein folding. New York : Academic Press, 1982. 6. Anfinsen CB, Haber E, Sela M, White FW. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci USA 1961; 47 : 1309–14. 7. Anfinsen CB. Principles that govern protein folding. Science 1973; 181 : 223–30. 8. Levinthal C. Are there pathways for protein folding ? J Chem Phys 1968; 65 : 44–5. 9. Tanford C. Protein denaturation. Adv Protein Chem 1968; 23 : 121–282. 10. Brandts JF. Thermodynamics of protein denaturation. J Am Chem Soc 1964; 86 : 4291–301 et 4302–14. 11. Privalov PL, Keshinaschvili NN. A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J Mol Biol 1974; 86 : 665–84. 12. Fersht AN. Nucleation mechanism in protein folding. Curr Opin Sruct Biol 1997; 7 : 3–9. 13. Baldwin RL. Intermediates in protein folding reactions and the mechanism of protein folding. Ann Rev Biochem 1975; 41 : 453–75. 14. Kim PS, Baldwin RL. Intermediate in the folding reactions of small proteins. Ann Rev Biochem 1990; 59 : 631–60. 15. Ptitsyn OB, Rashin AA. Stagewise mechanism of protein folding. Doklady Akademii Nauk SSSR 1973; 213 : 473–5. 16. Chothia C. Principles which determine the structure of proteins. Ann Rev Biochem 1974; 53 : 537–72. 17. Karplus M, Weaver DL. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci 1994; 3 : 650–68. 18. Dill KA. Theory for folding and stability of globular proteins. Biochemistry 1985; 24 : 1501–9. 19. Harrison SC, Durbin R. Is there a single pathway for the folding of a polypeptide chain ? Proc Natl Acad Sci
USA 1985; 82 : 4028–30. 20. Ikai A, Tanford C. Kinetics of unfolding and refolding of proteins. I. Mathematical analysis. J Mol Biol 1973; 73 : 145–54. 21. Hagerman PJ. Kinetic analysis of the reversible folding reactions of small proteins: application to the folding of lysozyme and cytochrome c. Biopolymers 1977; 16 : 731–47. 22. Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem 1995; 47 : 83–229. 23. Ogushi M, Wada A. Molten globule state: a compact form of protein with mobile side-chains. FEBS Lett 1983; 164 : 20–4. 24. Ptitsyn OB, Pain R, Semisotnov G, et al. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett 1990; 26 : 21–4. 25. Chaffotte AF, Cadieux C, Guillou Y, Goldberg ME. A possible folding intermediate: the C proteolytic domain of tryptophane synthase b chain folds in less than 4 milliseconds into a condensed state with non native-like secondary structures. Biochemistry 1992; 31 : 4303–8. 26. Arai M, Kuwajima K. Role of molten globule state in protein folding. Adv Protein Chem 2000; 57 : 209–82. 27. Pecorari F, Minard P, Desmadril M, Yon JM. Occurrence of transient multimeric species during the refolding of a monomeric protein. J Biol Chem 1996; 271 : 5270–6. 28.
Accounts of Chemical Research (special issue on protein folding) 1998; 11 (volume consacré aux résultats obtenus par les méthodes de cinétique ultrarapide). 29. Wolynes PG, Onuchic JN, Thuramalai D. Navigating the folding routes. Science 1995; 267 : 1618–20. 30. Yon JM. Aggregation, protein. In : Meyers RA, ed. Encyclopedia of molecular cell biology and molecular medicine. New York: John Wiley, 2004 : 23–52. 31. Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol 1997; 4 : 10–9. 32. Weil JH. Biochimie générale, 8e ed. Paris : Masson, 1997 : 31–2. |




